UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
WASHINGTON, D.C. 20549
FORM
(Mark One)
For
the fiscal year ended
OR
FOR THE TRANSITION PERIOD FROM TO
Commission
file number:
(Exact name of Registrant as specified in its charter)
(State or other jurisdiction of incorporation or organization) | (I.R.S. Employer Identification No.) |
(Address of principal executive offices, including zip code)
(Registrant’s telephone number, including area code)
Securities registered pursuant to Section 12(b) of the Act:
| Title of each class | Trading Symbol(s) | Name of each exchange on which registered | ||
Securities registered pursuant to Section 12(g) of the Act: N/A
Indicate
by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. Yes ☐
Indicate
by check mark if the registrant is not required to file reports pursuant to Section 13 or Section 15(d) of the Act.
Yes ☐
Indicate
by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the
Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required
to file such reports), and (2) has been subject to such filing requirements for the past 90 days.
Indicate
by check mark whether the registrant has submitted electronically every Interactive Data File required to be submitted pursuant
to Rule 405 of Regulation S-T (§232.405 of this chapter) during the preceding 12 months (or for such shorter period that
the registrant was required to submit such files).
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, a smaller reporting company or an emerging growth company. See the definitions of “large accelerated filer,” “accelerated filer,” “smaller reporting company” and “emerging growth company” in Rule 12b-2 of the Exchange Act:
| Large accelerated filer | ☐ | Accelerated filer | ☐ |
| ☒ | Smaller reporting company | ||
| Emerging growth company |
If an emerging growth company, indicate by check mark if the registrant has elected not to use the extended transition period for complying with any new or revised financial accounting standards provided pursuant to Section 13(a) of the Exchange Act. ☐
Indicate
by check mark whether the registrant has filed a report on and attestation to its management’s assessment of the effectiveness
of its internal control over financial reporting under Section 404(b) of the Sarbanes-Oxley Act (15 U.S.C. 7262(b)) by the registered
public accounting firm that prepared or issued its audit report.
Indicate
by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Act).
Yes ☐ No
The
aggregate market value of voting and non-voting common equity held by non-affiliates as of the last business day of the Registrant’s
most recently completed second fiscal quarter (June 30, 2021) was $
Documents Incorporated by Reference
The registrant has incorporated by reference into Part III of this Form 10-K portions of its Proxy Statement for its 2022 Annual Meeting of Shareholders. Such Proxy Statement will be filed with the Securities and Exchange Commission within 120 days of the registrant’s fiscal year ended December 31, 2021.
COHBAR, INC.
2021 FORM 10-K ANNUAL REPORT
Table of Contents
i
PART I
Forward-Looking Statements
This report, including the sections entitled “Business,” “Risk Factors” and “Management’s Discussion and Analysis of Financial Condition and Results of Operations,” contains forward-looking statements regarding future events and our future results that are based on our current expectations, estimates, forecasts and projections about our business, our results of operations, the industry in which we operate and the beliefs and assumptions of our management. Words such as “may,” “will,” “should,” “could,” “anticipate,” “believe,” “expect,” “intend,” “plan,” “potential,” “continue” and similar expressions are intended to identify these forward-looking statements. Examples of such forward-looking statements include statements regarding:
| ● | our future results of operations and financial position, business strategy, market size and potential growth opportunities; |
| ● | preclinical and clinical development activities; |
| ● | efficacy and safety profiles of our clinical candidates; |
| ● | the anticipated therapeutic properties of our drug development candidates; |
| ● | expectations regarding our ability to effectively protect our intellectual property; and |
| ● | expectations regarding our ability to attract and retain qualified employees and key personnel. |
These statements reflect our current beliefs and are based on information currently available to us. Forward-looking statements involve significant risks and uncertainties, including without limitation, those listed in the “Risk Factors” section. A number of factors could cause actual results to differ materially from the results discussed in the forward-looking statements including, but not limited to, changes in general economic and market conditions and the risk factors disclosed under “Risk Factors.” Although the forward-looking statements contained in this report are based upon what we believe to be reasonable assumptions, we cannot assure you that actual results will be consistent with these forward-looking statements. Investors should not place undue reliance on forward-looking statements. These forward-looking statements are made as of the date hereof and we assume no obligation to update or revise them to reflect new events or circumstances, except as required by applicable law.
Item 1. Business
OVERVIEW
CohBar (“CohBar,” “we,” “us,” “our,” “its” or the “Company”) is a clinical stage biotechnology company leveraging the power of the mitochondria and the peptides encoded in its genome to develop potential breakthrough therapeutics targeting chronic and age-related diseases with limited to no treatment options. Our novel approach is built on the key insights of the Company’s founders that certain mitochondrially encoded peptides produce effects that are not limited to local regulation within the mitochondria and may have important roles to play in critical systemic biological pathways. Many of these effects are quite distinct from traditional mitochondrial function such as energy production and metabolism, involving diverse processes including inflammation, fibrosis and cell signaling.
We believe we have achieved a leading position in exploring the mitochondrial genome and its utility for the development of novel therapeutics, including world-renowned expertise in mitochondrial biology, a broad intellectual property estate with more than 65 patent applications filed, key opinion leaders and disciplined drug discovery and development processes. Our proprietary processes of identifying nucleic acid sequences encoding native peptides in the mitochondrial genome, developing and optimizing novel analogs of these natural mitochondrial derived peptides (“MDPs”), as well as developing and conducting proprietary screens to identify and characterize the activities of these peptides are referred to as our Mito+ platform. We are using our Mito+ platform to identify and develop novel modified versions of natural peptides, which we call analogs, to treat a variety of serious conditions, with a focus on chronic diseases involving inflammation and fibrosis. We believe that the mitochondrial genome may be transformative in the field of drug discovery and that our novel peptide analogs may become a new and major class of drugs with broad therapeutic application. We are currently advancing a pipeline of novel peptide analogs through varying stages of development: CB5138-3 for idiopathic pulmonary fibrosis (“IPF”), CB4211 for the treatment of nonalcoholic steatohepatitis (“NASH”) and obesity, and several preclinical and discovery-stage programs.
1
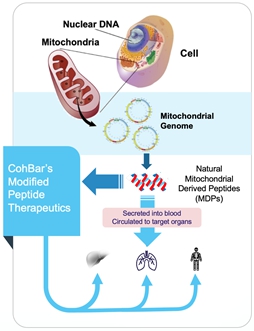
Potential of Mitochondrial Biology
Our approach leverages the longstanding symbiotic relationship between the mitochondria and the human cell, enabling us to take advantage of millennia of evolutionary pressure. While the central role of mitochondria as the powerhouse of the cell has been well understood for decades, recent research shows a much broader role for this important organelle. Mitochondria have been shown to signal within and between cells, orchestrate multiple biological systems, regulate metabolism and the immune system and control cell cycle, cell growth, and cell death (apoptosis). Other than the nucleus, the mitochondria are the only cell components that have their own genome and we believe the peptides encoded in the mitochondrial genome provide an effective starting point for the development of valuable therapeutics with the potential for better safety and tolerability profiles.
CohBar scientists have mined the mitochondrial genome and discovered multiple unique peptides. After creating novel analogs of these native peptides, we utilize a broad range of proprietary activity screens that are highly predictive of human activity and disease to assess the therapeutic potential of our novel peptides. Our novel analogs are then studied in in vitro and/or in vivo models to confirm their biological effects prior to the selection of a clinical candidate for further testing and ultimate entry into clinical trials. While we look to the mitochondrial genome as the source of our therapeutic peptides, we are not focused on relatively rare diseases caused by specific mitochondrial defects or abnormalities. Rather, our screening is geared towards detecting peptides that interact with cell surface receptors and have activity in important systemic biological pathways, resulting in product candidates with the potential to impact diseases with large unmet medical needs. Building on continued advances in our understanding of mitochondrial contributions to systemic processes, we have discovered a number of peptide families that are structurally unique and have distinct mechanisms of action, providing us with multiple independent opportunities for the successful development of novel therapeutics.
2
We believe that the proprietary capabilities of our Mito+ platform, combined with our scientific expertise and intellectual property portfolio, provide a competitive advantage in our mission to treat chronic and age-related diseases through the advancement of a new class of transformative drugs. Our peptide optimization process is designed to discover numerous potential drug candidate opportunities. These drug candidates may be internally developed by CohBar or advanced through strategic partnerships with larger biopharmaceutical companies. To ensure that we capture the most value from our pipeline, we aggressively file for broad intellectual property coverage, both in the United States and internationally, which we believe is critical to securing CohBar’s leadership role in the field and enabling us to benefit from prior and future discoveries.
We have filed more than 65 patent applications with claims directed to both compositions comprising and methods of using our novel MDPs and their analogs. We are the exclusive licensee from the Regents of the University of California and the Albert Einstein College of Medicine of additional patents that include claims that are directed to compositions comprising natural peptide sequences and their novel analogs and/or methods of their use in the treatment of indicated diseases.
Company Information
We were formed as a limited liability company in the state of Delaware in 2007 and converted to a Delaware corporation in 2009. We completed our initial public offering of common stock in January 2015 and our common stock is listed for trading on The Nasdaq Capital Market (CWBR).
Our corporate headquarters and laboratory are located in Menlo Park, California.
OUR STRATEGY
Our goal is to create a new and powerful class of medicines to address chronic and age-related diseases based on targeting the mitochondrial genome. The key elements of our strategy are to:
| ● | Focus our resources on developing therapies for high-value chronic and/or age-related diseases, with a focus on fibrotic and inflammatory conditions. |
| ● | Advance CB5138-3 through clinical development for IPF while continuing to evaluate additional potential indications. IPF is an area of high unmet medical need where we believe that CB5138-3 can offer differentiated advantages to patients and physicians. Given the broad anti-fibrotic and anti-inflammatory effects we have seen in our preclinical work, we also plan to evaluate other potential indications for this promising product candidate. |
| ● | Selectively form strategic alliances to augment our expertise in exploring the mitochondrial genome and its utility as a source of novel therapeutics to accelerate development and commercialization. We will continue to seek partners who can bring therapeutic expertise, development and commercialization capabilities and funding to allow us to maximize the potential of our pipeline. |
| ● | Leverage our Mito+ technology platform and unique peptide library to develop additional targets and programs. Our team has discovered multiple unique and previously unidentified peptides encoded within the mitochondrial genome that have benefitted from millennia of evolutionary pressure. We are evaluating the potential benefit of novel analogs of these peptides in a variety of models of fibrosis and inflammation, with the objective of maximizing the potential of our pipeline. |
3
OUR PIPELINE
Our research efforts are focused on utilizing our Mito+ platform to identify, assess and optimize novel analogs of native peptides found in the mitochondrial genome and advancing those candidates with the greatest therapeutic and commercial potential. Our pipeline includes a number of these novel peptide analogs in different stages of research and development.
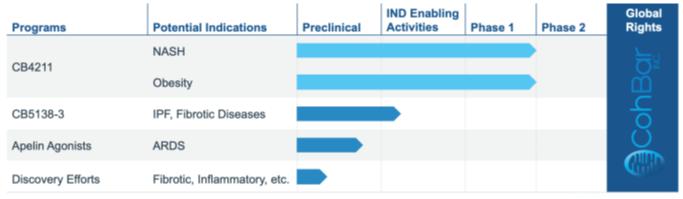
CB5138-3
In 2021, we nominated our second clinical candidate, CB5138-3, a first-in-class therapeutic under development for the treatment of idiopathic pulmonary fibrosis and other fibrotic diseases. Our CB5138-3 product candidate has impressive preclinical results, with significant anti-fibrotic and anti-inflammatory properties. In addition, we believe CB5138-3 has the potential to provide a better safety and tolerability profile than currently approved IPF drugs, which are poorly tolerated with significant gastrointestinal and/or skin toxicity. When combined with our promising preclinical data, we believe CB5138-3 could provide important clinical and commercial advantages over current standard of care. This program is currently in IND-enabling studies. To date, we have not seen any notable systemic toxicity in rodent or non-human primate studies. Due to additional planned formulation work, we plan to file an Investigational New Drug (“IND”) Application in the second half of 2023 and begin a first-in-human study shortly thereafter.
CB5138-3 Preclinical Studies
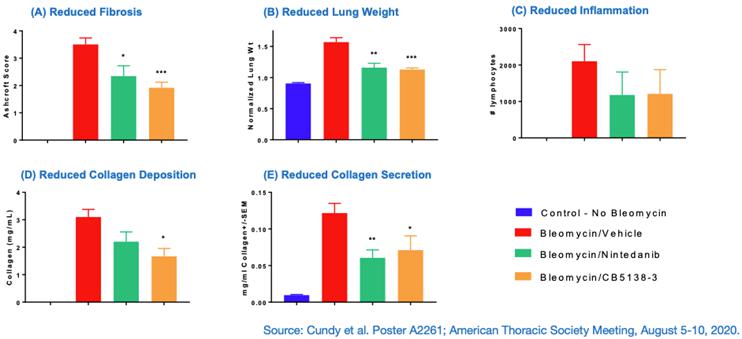
Multiple members of the CB5138 family of peptides have demonstrated anti-fibrotic and anti-inflammatory effects in in vitro and in vivo models. For example, in co-cultures of human lung cells, CB5138-1, a peptide closely related to CB5138-3, decreased the expression of key fibrosis biomarkers, including alpha smooth muscle actin (αSMA), and collagen types I and III. CB5138-1 also decreased the transformation of healthy lung cells into fibrotic cells after induction by TGF-beta1, resulting in reduced production of the fibrotic components αSMA and pro-collagen I alpha 1. Using the therapeutic mouse model of IPF, where peptide treatment is started one week after fibrosis induction with bleomycin, CB5138-3 significantly reduced lung fibrosis assessed by the Ashcroft Score, reduced inflammation, and decreased fibrosis-related changes in lung weight, collagen deposition in lung tissue, and collagen secretion into lung fluid. Data from these studies were presented at the American Thoracic Society (ATS) 2020 Meeting.
4
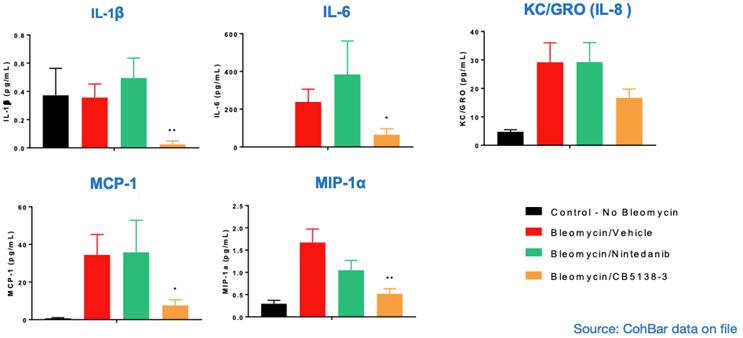
In the same therapeutic mouse model of IPF, CB5138-3 demonstrated significant favorable anti-inflammatory effects, as evidenced by a reduction in various pro-inflammatory cytokines, chemokines and inflammatory cells in lung fluid (BALF).
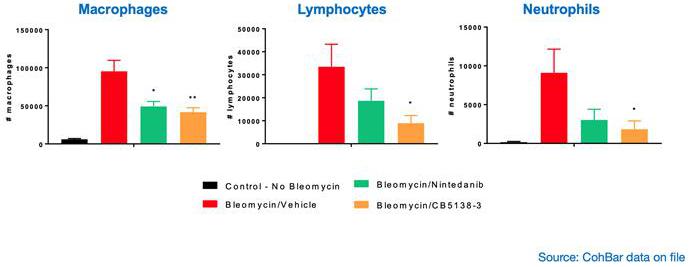
5
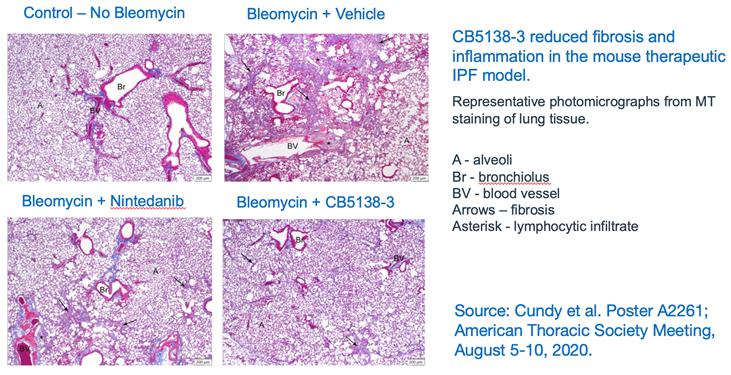
As seen through the staining of lung tissue, the data from the therapeutic mouse model of IPF demonstrated that CB5138-3 reduced fibrosis and inflammation.
CB4211
Our most advanced clinical candidate, CB4211, is a first-in-class therapeutic under development for the treatment of NASH and obesity. CB4211 recently demonstrated positive effects on reducing biomarkers of liver injury and improving metabolic homeostasis in a Phase 1a/1b clinical study in obese subjects with nonalcoholic fatty liver disease (“NAFLD”). CB4211 is a novel and improved analog of MOTS-c, a naturally occurring MDP. MOTS-c was discovered in 2012 by CohBar founder Dr. Pinchas Cohen and his academic collaborators and has been shown to play a significant role in the regulation of metabolism in animal models. Compared to other assets under development for the treatment of NASH, CB4211 has a unique mechanism of action, which we believe offers a differentiated approach to treating NASH and obesity, as well as the potential to exhibit an enhanced safety profile due to its natural origin. Furthermore, we believe the positive clinical data from our CB4211 trial is an important validation of our overall approach to drug discovery, serving as a proof point that novel analogs of peptides encoded in the mitochondrial genome can impact systemic biological pathways in humans while having an attractive safety and tolerability profile. We have been working to further improve the formulation for CB4211 and intend to partner this program before moving forward into further clinical trials.
CB4211 Mechanism of Action
We have shown that CB4211 has impacts on regulating fatty acid metabolism, glucose homeostasis, and insulin sensitivity. These studies demonstrated that CB4211 potentiates insulin effects on fatty acid metabolism and glucose homeostasis by extending the duration of insulin receptor (“IR”) activation without altering the magnitude of the response or activation of highly related receptors. For example, CB4211 potentiated insulin-mediated inhibition of lipolysis in isoproterenol-stimulated adipocyte cultures without changing maximal response, while CB4211 alone had no effect. Subsequent de-phosphorylation of IR and downstream targets (IRS-1 and Akt) was markedly slowed in the presence of insulin with CB4211 compared to insulin alone. Inhibitors of IR auto-phosphorylation (GSK183705A) or downstream PI3K/Akt signaling pathway components (wortmannin, Akti-1/2) abolished the antilipolytic effects of insulin alone and in combination with CB4211. Further supporting specificity of insulin signaling, CB4211 enhanced insulin-mediated phosphorylation of IR, IRS-1, and Akt, without affecting IGF mediated phosphorylation of IGF-1R. Consistent with activity through the IR, CB4211 potentiated insulin-induced reduction in glucose production in H4-IIE cells. The acute in vivo effect of CB4211 on insulin tolerance was determined in fasted DIO mice. Administration of CB4211 with insulin enhanced insulin sensitivity, prolonging the reduction in blood glucose levels compared to insulin alone. Data from these studies were presented at the American Diabetes Association (“ADA”) 2018 Meeting.
6
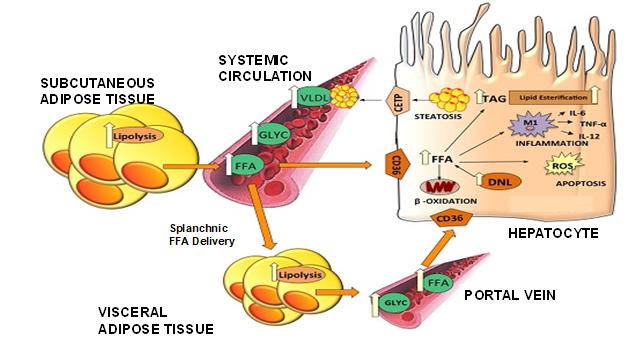
Source: Nutrients 2015,7, 9453–9474
Summary of Results from Phase 1a/1b Clinical Study of CB4211 in Obese Subjects with NAFLD
In August 2021, we released positive topline data from our Phase 1a/1b clinical study of CB4211. The Phase 1a stage of the study was designed to assess the safety, tolerability, and pharmacokinetics of CB4211 following single and multiple-ascending doses in healthy subjects. Subjects in the Phase 1a study experienced mild, but persistent injection site reactions, which were generally seen as painless bumps at the injection site that can be felt under the skin, but in most cases would be otherwise undetectable. We modified the formulation for CB4211 partway through the Phase 1a study and did not observe any persistent injection site bumps with the modified formulation. The subsequent Phase 1b stage was designed to assess the safety, tolerability, and activity of CB4211 in obese subjects with NAFLD. The study met its primary endpoint as CB4211 was well-tolerated and appeared safe with no serious adverse events. The evaluation of the exploratory endpoints in the Phase 1b portion of the trial showed significant reductions from baseline in key biomarkers of liver damage, ALT and AST, and in glucose levels in the CB4211 group compared to placebo after four weeks of treatment, with a trend towards lower body weight. Data from the study were presented at the American Association for the Study of Liver Disease (AASLD) 2021 Liver Meeting®.
Key findings from the topline data of the Phase 1b portion of the study are summarized below.
| Biomarker |
CB4211 (25 mg) (n = 11) |
Placebo (n = 9) |
Difference from Placebo |
MRI-PDFF Data |
CB4211 (25 mg) (n = 11) |
Placebo (n = 9) | |
|
ALT (% reduction from baseline) |
-21% | 4% | -25* |
Baseline Liver Fat Content (LFC) |
21.1% | 15.9% | |
|
Proportion of subjects with >17 U/L decrease in ALT(1) |
27% | 11% | 16% |
Percent Reduction in LFC (Absolute) |
-5.03% |
-4.88% | |
|
AST (% reduction from baseline) |
-28% | -11% | -17%* | Proportion of Responders Achieving >30% Relative Reduction in LFC(2) | 36% | 33% | |
|
Glucose (% reduction from baseline) |
-6% | 0% | -6%* | ||||
ALT: Alanine aminotransferase. AST: Aspartate aminotransferase.
| * | Statistically significant versus placebo, p<0.05 by unpaired t test |
| (1) | A decrease in ALT by 17 U/L or more is significantly associated with histologic response in NASH (Loomba R et al. Gastroenterology, 2019; 156 (1): 88-95) |
MRI-PDFF: Magnetic resonance imaging – proton density fat fraction.
| (2) | A relative reduction of 30% in liver fat is associated with a histological response in non-alcoholic steatohepatitis (Patel J et al. Therap Adv Gastroenterol 2016, 9(5): 692-701) |
7
CB4211 Preclinical Studies
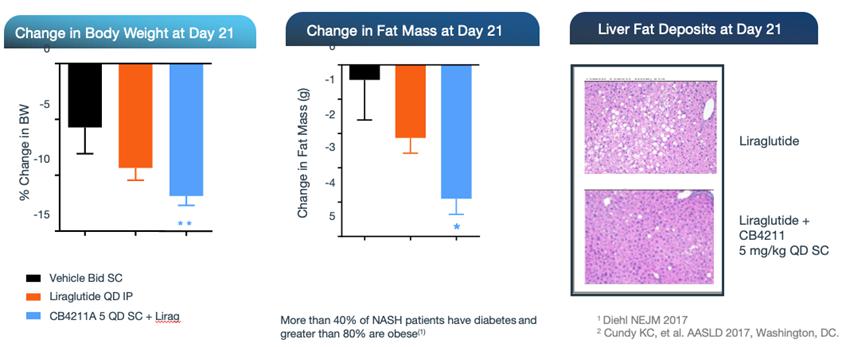
In preclinical studies, CB4211 demonstrated significant therapeutic potential for the treatment of NASH, showing improvements in triglyceride levels, as well as favorable effects on liver enzyme markers associated with NAFLD and NASH. CB4211 also demonstrated significant therapeutic potential for the treatment of obesity, demonstrating significantly greater weight loss together with more selective reduction of fat mass versus lean mass in comparison to the GLP-1 agonist liraglutide, the active ingredient in a market-leading obesity drug, in DIO mice. The therapeutic effects of CB4211 have been further evaluated in the well-established Stelic Animal Model (STAM™) of NASH. In this model, treatment with CB4211 resulted in a significant reduction of the non-alcoholic fatty liver disease activity score, or NAS, a composite measure of steatosis (fat accumulation), inflammation and hepatocyte ballooning (cellular injury). Data from these studies were presented at the AASLD 2017 Liver Meeting®.
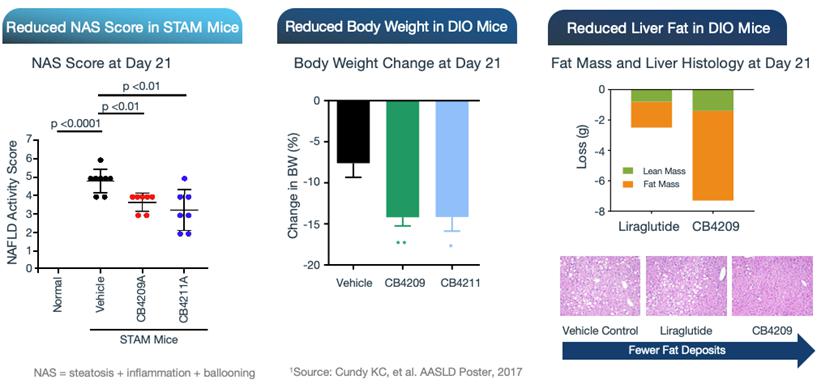
In addition, in a mouse model of NASH, CB4211 demonstrated a synergistic effect with liraglutide, with the combination resulting in a significant reduction in body weight and fat mass, as well as a decrease in liver fat, in obese mice.
Discovery Efforts
Our discovery efforts have resulted in the identification of multiple unique and previously unidentified peptides encoded within the mitochondrial genome. Many of these natural sequences and their novel analogs have demonstrated various degrees of biological activity in cell based and/or animal models relevant to a wide range of diseases. Our research efforts have identified and focused on certain of these novel analogs that have demonstrated greatest therapeutic potential. We plan to further explore these peptide families for the potential treatment of a variety of diseases, subject to resource availability and the requirements of our more-advanced programs.
8
CB5064 Analogs
Our discovery efforts have identified CB5064 Analogs, a family of peptides that are agonists of the apelin receptor. By utilizing the protective apelin signaling pathway, our CB5064 Analogs have the potential to address a variety of unmet medical needs such as our initial target of Acute Respiratory Distress Syndrome (“ARDS”). We believe our CB5064 Analogs could be effective in ARDS from a variety of different causes, such as bacterial or viral pneumonia, including COVID-19 associated ARDS. In a preclinical mouse model of ARDS, treatment with CB5064 Analogs reduced fluid accumulation in the lungs and a corresponding broad reduction in levels of key pro-inflammatory cytokines secreted into the lung fluid, when compared to treatment with a placebo control.
CB5064 Analogs Mechanism of Action
Apelin is an endogenous peptide produced and secreted by several cell types, including fat (adipose tissue) and muscle cells, that activates the apelin receptor (“APJ”), a key cell surface receptor. The apelin/APJ axis is involved in protective regulation of fluid homeostasis, cardiovascular function, and metabolism. Activation of APJ, which is broadly expressed but particularly abundant in pulmonary and cardiac tissues, is known to be protective in animal models of ARDS, thrombosis, stroke and sepsis. In addition to its protective effects in lung injury, apelin has also been shown to reduce body weight and improve insulin sensitivity in obese mice. Apelin itself is a poor drug candidate due to its relative instability and short half-life.
Disease Focus
Our research and development focus is predominantly on chronic and age-related diseases and our lead programs are targeted to the following conditions.
Idiopathic Pulmonary Fibrosis – Idiopathic Pulmonary Fibrosis is a chronic, progressive, debilitating, and usually fatal interstitial lung disease that affects approximately 100,000 people in the United States This orphan disease results in fibrosis of the lungs. Idiopathic means “of unknown cause,” though there are certain risk factors that are associated with a higher incidence of IPF, including age (> 50), male gender, smoking, acid reflux and family history of IPF. While many patients do not have symptoms early in the course of the disease, as IPF progresses, symptoms can include persistent dry cough, shortness of breath, especially with exertion, chest pain, loss of appetite and non-intentional weight loss, fatigue and swelling in the legs. Mean survival after diagnosis is only two to three years, which is worse than many cancers. There are two FDA-approved drugs to treat IPF. While both drugs have been shown to decrease the rate of loss of lung function, neither has demonstrated an improvement in survival and both are poorly tolerated by many patients.
NASH – NAFLD is the build-up of extra fat in liver cells that is not due to alcohol consumption. In some patients, NAFLD leads to NASH, a progressive condition involving inflammation and ultimately fibrosis, or scarring, of the liver. This can further progress to cirrhosis (advanced, late-stage scarring). Patients who develop cirrhosis are at risk for complications including liver failure and liver cancer (hepatocellular carcinoma). While the cause of NASH is unknown, it is associated with a broader set of metabolic disorders and important risk factors include elevated triglyceride or cholesterol levels, type 2 diabetes, high blood pressure and obesity, particularly with body fat concentrated around the waist. NASH is also more prevalent in certain ethnic groups including Asian and Hispanic populations. Since there are generally no symptoms until late in the disease progression, many patients have significant liver damage by the time the diagnosis is made. NASH is estimated to impact approximately 12% of the U.S. adult population and there is currently no approved treatment.
Obesity – Obesity impacts more than 40% of U.S. adults and is a major risk factor for a variety of other serious diseases, including heart disease, stroke, type 2 diabetes, NASH and certain types of cancer. Lifestyle interventions have limited success and there is a growing recognition of the need for safe and effective treatments for this important metabolic condition.
Acute Respiratory Distress Syndrome – ARDS occurs when fluid builds up in the tiny, elastic air sacs, or alveoli, of the lungs, resulting in poor blood oxygenation. ARDS is typically a complication of some other primary condition, such as pneumonia, sepsis, or trauma. Current treatment is primarily supportive, along with treatment of the underlying infection or trigger. Mortality rates are high and many patients that survive experience lasting lung damage.
9
COMPETITION
The biopharmaceutical industry is characterized by rapidly advancing technologies, intense competition and a strong emphasis on proprietary products. While we believe that our scientific knowledge, technology, and research and development experience provide us with competitive advantages, we face potential competition from many different sources, including major biopharmaceutical, specialty pharmaceutical and biotechnology companies, academic institutions and governmental agencies, and public and private research institutions. Many of our competitors may have significantly greater financial resources and capabilities for research and development, manufacturing, preclinical testing, conducting clinical trials, obtaining regulatory approvals and marketing approved products than we do. Although our product candidates have unique mechanisms of action compared to most other approved or investigational therapies across the disease areas where we are focusing our development, we will need to compete with currently approved therapies, and potentially those currently in development if they are approved. We are aware of several marketed and investigational products in our leading disease areas, including but not limited to:
| ● | IPF: There are two FDA approved drugs to treat IPF: nintedanib (Ofev), marketed by Boehringer Ingelheim GmbH, and pirfenidone (Esbriet), marketed by Roche Holdings AG. In addition, there are several companies developing product candidates to treat IPF, including AbbVie, Boehringer Ingelheim GmbH, FibroGen, Inc., Galecto, Inc., Pliant Therapeutics, Inc. and Roche Holdings AG. |
| ● | NASH: There are currently no approved therapies for the treatment of NASH. There are several companies developing product candidates to treat NASH, including Madrigal Pharmaceuticals, Inc., Intercept Pharmaceuticals, Inc., Novo Nordisk, Pfizer Inc., Gilead Sciences, Inc., AstraZeneca plc, Eli Lilly & Company, GlaxoSmithKline plc, Amgen, Inc., BMS, Johnson & Johnson, Merck & Co., Inc., Roche Holdings AG, Viking Therapeutics, Inc., Akero Therapeutics, Inc. and Hepion Pharmaceuticals, Inc. |
| ● | Obesity: There are several products currently approved for obesity, such as Saxenda, Contrave, Wegovy, phentermine (Adipex) and other sympathomimetic amines approved for short term use (a few weeks) such as benzphetamine (Didex), diethyoproprion (Tenuate) and phendimetrazine (Bontril), Xenical and Alli, and Qsymia, as well as several investigational therapies that are currently being studied for the treatment of obesity. |
| ● | ARDS: There are no FDA approved drugs to specifically treat ARDS. There are several companies developing product candidates to treat ARDS, including Biohaven Pharmaceutical Holding Company Ltd., Boehringer Ingelheim GmbH, Faron Pharmaceuticals Ltd. Athersys, Inc., and Edesa Biotech. |
EMPLOYEES AND HUMAN CAPITAL RESOURCES
As of March 24, 2022, we had ten employees, nine full-time and one part-time. Additionally, from time to time we engage subject-matter experts on a consulting basis in specific areas of our research and development efforts. None of our employees are represented by a labor union or covered by a collective bargaining agreement. We have not experienced any work stoppages and we consider our relations with our employees to be good.
Our human capital resources objectives include, as applicable, identifying, recruiting, retaining, incentivizing and integrating our existing and additional employees. The principal purposes of our equity incentive plans are to attract, retain and motivate selected employees, consultants and directors through the granting of stock-based compensation awards and cash-based performance bonus awards.
RESEARCH AND DEVELOPMENT
Research and development activities are central to our business model. Our research programs include activities related to discovery of novel MDPs, investigational research to evaluate the potential therapeutic effects of certain discovered natural sequences in research and preclinical studies and engineering novel, improved analogs of certain discovered natural sequences with characteristics suitable for further development as potential drug candidates and advancing our identified candidates through clinical studies. Depending on factors of capability, cost, efficiency and intellectual property rights, we conduct our research programs independently at our laboratory facility. We also outsource some research and development activities pursuant to contractual arrangements with CROs or under collaborative arrangements with academic institutions.
10
INTELLECTUAL PROPERTY
Patents
Our commercial success depends in part on our ability to obtain and maintain proprietary protection for our novel biological discoveries and therapeutic methods, to operate without infringing on the proprietary rights of others and to prevent others from infringing our proprietary rights. We seek to protect our proprietary position by, among other methods, licensing and/or filing patent applications related to our proprietary technology, inventions and improvements that are important to the development and implementation of our business.
Our intellectual property and patent strategy is focused on our MDPs and our novel analogs of these natural peptides. Our strategy is generally to seek patent protection in the United States and, where applicable, in those international jurisdictions we identify as holding significant potential market opportunity for any drug we may develop and in which patent protection is available. We also rely on trade secrets, know-how, continuing technological innovation and potential in-licensing opportunities to develop and maintain our proprietary position. With respect to new biologically active MDPs that we identify within the mitochondrial genome, we typically file provisional patent applications and seek composition-of-matter and method-of-treatment patents for our MDPs, and/or their novel analogs, and prospective novel drug candidates as well as methods of use based on research and preclinical evaluation of therapeutic potential. We intend to file international Patent Cooperation Treaty (“PCT”) applications and/or non-provisional patent applications for those MDPs and/or novel analogs within our pipeline based on further assessment of their therapeutic and commercial potential, as well as strategic and competitive considerations. We believe that the opportunity to engineer analogs or create combination therapies will afford us the opportunity to strengthen IP protection for our drug development candidates as they advance through our development pipeline and to broaden our IP protection internationally.
As of December 31, 2021, we have filed more than 65 patent applications, including at least 10 international PCT applications, with claims directed to both composition of matter and methods of use of novel MDPs and their novel analogs. Our patent applications include filings in the United States, Europe and a number of other foreign countries, with projected expiration dates ranging from 2037 to 2041. Additionally, we are the exclusive worldwide licensee from the Regents of the University of California (the “Regents”) of 15 issued patents that will expire between 2028 and 2034. Other licensed intellectual property is described below.
Terms for individual patents extend for varying periods of time generally depending on the date of filing of the patent application and the legal term of patents in the countries in which they are obtained. Generally, patents issued from applications filed in the United States are effective for twenty years from the earliest non-provisional filing date. In addition, in certain instances, a patent term can be extended to recapture a portion of the term effectively lost as a result of the FDA regulatory review period; however, the restoration period cannot be longer than five years and the total patent term, including the restoration period, must not exceed fourteen years following FDA approval. The duration of foreign patents varies in accordance with provisions of applicable local law, but typically is also twenty years from the earliest international filing date. In certain instances, extension of patent term due to regulatory approval activities is available in foreign countries.
National and international patent laws concerning peptide therapeutics remain highly unsettled. Policies regarding the patent eligibility or breadth of claims allowed in such patents are currently in flux in the United States and other countries. Changes in either the patent laws or in interpretations of patent laws in the United States and other countries can diminish our ability to protect our inventions and enforce our intellectual property rights. Accordingly, we cannot predict the breadth or enforceability of claims that may be granted in our patents or in third-party patents. The biopharmaceutical industry is characterized by extensive litigation regarding patents and other intellectual property rights. Our ability to maintain and solidify our proprietary position for our drugs and technology will depend on our success in obtaining effective claims and enforcing those claims once granted. We do not know whether any of the patent applications that we may file or license from third parties will result in the issuance of any patents. The issued patents that we own, license, or may license or own in the future, may be challenged, invalidated or circumvented, and the rights granted under any issued patents may not provide us with sufficient protection or competitive advantages against competitors with similar technology. Furthermore, our competitors may be able to independently develop and commercialize similar drugs or duplicate our technology, business model or strategy without infringing our patents. Because of the extensive time required for clinical development and regulatory review of a drug we may develop, it is possible that, before any of our drugs can be commercialized, any related patent may expire or remain in force for only a short period following commercialization, thereby reducing any advantage of any such patent.
Summaries of our owned and licensed patent positions are described below.
11
CohBar Owned IP
As of December 31, 2021, we have filed more than 65 patent applications, including applications relating to CB4211, CB5138 Analogs and other CohBar-identified MDPs and novel analogs.
MOTS-c Analog Patent Coverage
We have filed over 20 U.S. and foreign patent applications, including applications in Europe and Asian countries, directed to novel refined analogs of MOTS-c with improved properties, including claims directed to composition of matter and methods of use as well as to formulations containing these peptides. These applications also cover our most advanced product candidate CB4211 and its formulations. If issued, these patents would expire in 2037 or 2039. In 2021, the U.S. Patent and Trademark Office (“USPTO”) granted us a patent that covers CB4211 and related compositions, as well as methods of treatment, including methods of treating NASH. The term of this patent extends to 2037.
CB5138 Analog Patent Coverage
We have filed national and regional patent applications related to a CohBar-identified MDP (CB5138) and novel, improved analogs, including claims directed to composition of matter and methods of use, with a projected expiration date of 2040.
Other CohBar Identified MDPs and Analog Coverage
We have also filed more than 45 provisional patent applications and at least 10 PCT applications related to additional CohBar-identified MDPs and/or their novel, improved analogs, including claims directed to compositions of matter and methods of use. A number of these filings relate to our preclinical programs, including our CB5064 analogs. We intend to file additional non-provisional U.S. patent applications and/or other regional or national patent application for MDPs and/or novel analogs within our pipeline based on further assessments of their therapeutic and commercial potential, as well as strategic and competitive considerations.
CohBar Licensed IP
MOTS-c Patent Coverage
We are the exclusive licensee from the Regents to intellectual property rights related to MOTS-c, including two issued U.S. patents as well as corresponding foreign applications and granted foreign patents filed in multiple countries and regions. These issued patents and applications include composition of matter claims directed to MOTS-c and certain novel analogs of MOTS-c, as well as methods of use claims for MOTS-c or certain novel analogs of MOTS-c as a treatment for type 1 diabetes, T2D, fatty liver, obesity and cancer. Patents related to these filings have been granted in the United States, Europe, Japan and several other countries.
SHLP-2 and SHLP-6 Patent Coverage
We are the exclusive licensee from the Regents to intellectual property for SHLP-2 and SHLP-6 and their novel analogs. This intellectual property includes an issued U.S. patent with a term expiring in 2029.
Humanin and Humanin Analogs Patent Coverage
We are the exclusive licensee from the Regents and the Albert Einstein College of Medicine of Yeshiva University of two U.S. issued patents covering humanin and humanin analogs for treatment of disease which expire in 2028 and 2029.
Trade Secrets
In addition to patents, we rely upon unpatented trade secrets, know-how and continuing technological innovation to develop and maintain our competitive position. We seek to protect our proprietary information, in part, using confidentiality agreements with our commercial partners, collaborators, employees and consultants and invention assignment agreements with our employees. These agreements are designed to protect our proprietary information and, in the case of the invention assignment agreements, to grant us ownership of technologies that are developed through a relationship with a third party. These agreements may be breached, and we may not have adequate remedies for any breach. In addition, our trade secrets may otherwise become known or be independently discovered by competitors. To the extent that our commercial partners, collaborators, employees and consultants use intellectual property owned by others in their work for us, disputes may arise as to the rights in related or resulting know-how and inventions.
12
Trademarks
We consider COHBARTM to be our common law trademark and are pursuing registration in the United States Patent & Trademark Office. We are also pursuing trademark registration for MITO+ for use in conjunction with research and development of pharmaceutical products.
In-licenses
MOTS-c Exclusive License
On August 6, 2013, we entered into an exclusive license agreement with the Regents to obtain worldwide, exclusive rights under patent filings and other intellectual property rights in inventions developed by Dr. Cohen and academic collaborators at the University of California, Los Angeles. The intellectual property includes the U.S. and foreign patents and patent applications described above under “MOTS-c Patent Coverage.”
We agreed to pay the Regents specified development milestone payments aggregating up to $765,000 for the first product sold under the license. Milestone payments for additional products developed and sold under the license are reduced by 50%. We are also required to pay annual maintenance fees to the licensors. Aggregate maintenance fees for the first three years following execution of the agreement were $7,500. Thereafter, we are required to pay maintenance fees of $5,000 annually until the first sale of a licensed product. In addition, we are required to pay the Regents royalties equal to 2% of our worldwide net sales of drugs, therapies or other products developed from claims covered by the licensed patent, subject to a minimum royalty payment of $75,000 annually, beginning after the first commercial sale of a licensed product. We are required to pay the Regents royalties ranging from 8% of worldwide sublicense sales of covered products (if the sublicense is entered after commencement of Phase II clinical trials) to 12% of worldwide sublicense sales (if the sublicense is entered prior to commencement of Phase I clinical trials). The agreement also requires us to meet certain diligence and development milestones, including filing of an IND Application for a product covered by the agreement on or before the seventh anniversary of the agreement date.
Under the agreement, the license rights granted to us are subject to any rights the U.S. government may have in such licensed rights due to its sponsorship of research that led to the creation of the licensed rights. The agreement also provides that if the Regents become aware of a third-party’s interest in exploiting the licensed technologies in a field that we are not actively pursuing, then we may be obligated either to issue a sublicense for use in the unexploited field to the third-party on substantially similar terms or to actively pursue the unexploited field subject to appropriate diligence milestones. The agreement terminates upon the expiration of the last valid claim of the licensed patent rights. We may terminate the agreement at any time by giving the Regents advance written notice. The agreement may also be terminated by the Regents in the event of our continuing material breach after notice of such breach and the opportunity to cure.
Humanin and SHLPs Exclusive License
On November 30, 2011, we entered into an exclusive license agreement with the Regents and the Albert Einstein College of Medicine at Yeshiva University to obtain worldwide, exclusive rights under patent filings and other intellectual property rights in inventions developed by Drs. Cohen and Barzilai and their academic collaborators. The intellectual property includes the U.S. patents described above under “Humanin and Humanin Analogs Patent Coverage” and “SHLP-2 and SHLP-6 Patent Coverage.”
We agreed to pay the licensors specified development milestone payments aggregating up to $765,000 for the first product sold under the license. Milestone payments for additional products developed and sold under the license are reduced by 50%. We are also required to pay annual maintenance fees to the licensors. Aggregate maintenance fees for the first five years following execution of the agreement were $80,000. Thereafter, we are required to pay maintenance fees of $50,000 annually until the first sale of a licensed product. In addition, we are required to pay the licensors royalties equal to 2% of our worldwide net sales of drugs, therapies or other products developed from claims covered by the licensed patents, subject to a minimum royalty payment of $75,000 annually, beginning after the first commercial sale of a licensed product. We are required to pay royalties ranging from 8% of worldwide sublicense sales of covered products (if the sublicense is entered after commencement of Phase II clinical trials) to 12% of worldwide sublicense sales (if the sublicense is entered prior to commencement of Phase I clinical trials). The agreement also requires us to meet certain diligence and development milestones, including filing of an IND for a product covered by the agreement on or before the seventh anniversary of the agreement date.
Under the agreement, the license rights granted to us are subject to any rights the U.S. government may have in such licensed rights due to its sponsorship of research that led to the creation of the licensed rights. The agreement terminates upon the expiration of the last valid claim of the licensed patent rights. We may terminate the agreement at any time by giving the Regents advance written notice. The agreement may be modified or terminated on a product-by-product basis by the Regents if we materially fail to meet certain diligence requirements and development milestones. The agreement may also be terminated by the Regents in the event of our continuing material breach after notice of such breach and the opportunity to cure. In October 2021, the Regents accepted our payment for an additional year of license maintenance.
13
ENVIRONMENTAL AND OTHER REGULATORY MATTERS
Government Regulation
The preclinical studies and clinical testing, manufacture, labeling, storage, record keeping, advertising, promotion, export, marketing and sales, among other things, of our therapeutic candidates and future products, are subject to extensive regulation by governmental authorities in the United States and other countries. In the United States, pharmaceutical products are regulated by the Food and Drug Administration (the “FDA”) under the Federal Food, Drug, and Cosmetic Act (the “FDCA”) and other laws. Biologics are subject to regulation by the FDA under the FDCA, the Public Health Service Act, and related regulations, and other federal, state and local statutes and regulations. Biological products include, among other things, viruses, therapeutic serums, vaccines and most protein products. Product development and approval within these regulatory frameworks takes a number of years, and involves the expenditure of substantial resources.
Regulatory approval will be required in all major markets in which we, or our licensees, seek to test our products in development. At a minimum, such approval requires evaluation of data relating to quality, safety and efficacy of a product for its proposed use. The specific types of data required and the regulations relating to these data differ depending on the territory, the drug involved, the proposed indication and the stage of development.
In general, new chemical entities are tested in animal models to determine whether the product is reasonably safe for initial human testing. Additional preclinical testing continues during the clinical development stage. Clinical trials for new products are typically conducted in three sequential phases that may overlap. Phase 1 trials typically involve the initial introduction of the pharmaceutical into healthy human volunteers and focus on testing for safety, dosage tolerance, metabolism, distribution, excretion and clinical pharmacology. In the case of serious or life-threatening diseases, such as cancer, initial Phase 1 trials are often conducted in patients directly, with preliminary exploration of potential efficacy. Phase 2 trials involve clinical trials to evaluate the effectiveness of the drug for a particular disease indication or indications in patients with the disease or condition under study and to determine appropriate dosages and dose regimens and the common short-term side effects and risks associated with the drug. Phase 2 trials are typically closely monitored and conducted in a relatively small number of patients, usually involving no more than several hundred subjects. Phase 3 trials are generally expanded, well-controlled clinical trials. They are performed after preliminary evidence suggesting effectiveness, as well as the appropriate dose and dose ranges of the drug, have been obtained, and are intended to gather the additional information about effectiveness and safety that is needed to evaluate the overall benefit-risk relationship of the drug and to provide an adequate basis for product labeling.
In the United States, specific research and preclinical data, chemical data and a proposed clinical study protocol, as described above, must be submitted to the FDA as part of an Investigational New Drug application, or IND, which, unless the FDA objects, will become effective 30 days following receipt by the FDA. Phase 1 trials may commence only after the IND application becomes effective. Following completion of Phase 1 trials, further submissions to regulatory authorities are necessary in relation to Phase 2 and 3 trials to update the existing IND. Authorities may require additional data before allowing the trials to commence and could demand discontinuation of studies at any time if there are significant safety issues. In addition to regulatory review, a clinical trial involving human subjects has to be approved by an independent body. The exact composition and responsibilities of this body differ from country to country. In the United States, for example, each clinical trial is conducted under the auspices of an Institutional Review Board for any institution at which the clinical trial is conducted. This board considers among other factors, the design of the clinical trial, ethical factors, the safety of the human subjects and the possible liability risk for the institution.
14
Information generated in this process is susceptible to varying interpretations that could delay, limit, or prevent regulatory approval at any stage of the approval process. Failure to demonstrate adequately the quality, safety and efficacy of a therapeutic drug under development would delay or prevent regulatory approval of the product.
In order to gain marketing approval, we must submit a new drug application, or NDA, for review by the FDA. The NDA must include a substantial amount of data and other information concerning safety and effectiveness of the drug compound from laboratory, animal and clinical testing, as well as data and information on manufacturing, product stability, and proposed product labeling.
There can be no assurance that if clinical trials are completed that we or any future collaborative partners will submit an NDA or similar applications outside of the United States for required authorizations to manufacture or market potential products, or that any such applications will be reviewed or approved in a timely manner. Approval of an NDA, if granted at all, can take several months to several years, and the approval process can be affected by a number of factors. Additional studies or clinical trials may be requested during the review and may delay marketing approval and involve unbudgeted costs. Regulatory authorities may conduct inspections of relevant facilities and review manufacturing procedures, operating systems and personnel qualifications. In addition to obtaining approval for each product, in many cases each drug manufacturing facility must be approved. Further, inspections may occur over the life of the product. An inspection of the clinical investigation sites by a competent authority may be required as part of the regulatory approval procedure. As a condition of marketing approval, the regulatory agency may require post-marketing surveillance to monitor adverse effects, or other additional studies as deemed appropriate. After approval for the initial disease indication, further clinical studies are usually necessary to gain approval for additional indications. The terms of any approval, including labeling content, may be more restrictive than expected and could affect product marketability.
Holders of an approved NDA are required to report certain adverse reactions and production problems, if any, to the FDA, and to comply with certain requirements concerning advertising and promotional labeling for their products. Moreover, quality control and manufacturing procedures must continue to conform to current good manufacturing practices (“cGMP”) after approval, and the FDA periodically inspects manufacturing facilities to assess cGMP compliance. Accordingly, manufacturers must continue to expend time, money and effort in the area of production and quality control to maintain compliance with cGMP and other aspects of regulatory compliance. We expect to continue to rely upon third-party manufacturers to produce commercial supplies of any products which are approved for marketing. We cannot be sure that those manufacturers will remain in compliance with applicable regulations, or that future FDA inspections will not identify compliance issues at the facilities of our contract manufacturers that may disrupt production or distribution, or require substantial resources to correct.
Any of our future products approved by the FDA will likely be purchased principally by patients through a pharmacy benefit plan or by pharmacies that typically bill various third-party payers, such as governmental programs (e.g., Medicare and Medicaid), private insurance plans and managed care plans, for the pharmaceuticals provided to patients. The ability of customers to obtain appropriate reimbursement for the products they purchase is crucial to the success of new drug and biologic products. The availability of reimbursement affects which products customers purchase and the prices they are willing to pay. Reimbursement varies from country to country and can significantly impact the acceptance of new products. Even if we were to develop a promising new product, we may find limited demand for the product unless reimbursement approval is obtained from private and governmental third-party payers.
15
In the United States and some foreign jurisdictions, there have been a number of legislative and regulatory changes and proposed changes regarding the health care system and efforts to control health care costs, including drug prices, that could significantly affect the development of our business, including preventing, limiting or delaying regulatory approval of our drug candidates and reducing the sales and profits derived from our products once they are approved. For example, in the United States, the Patient Protection and Affordable Care Act of 2010 (“ACA”) substantially changed the way health care is financed by both governmental and private insurers and significantly affects the biopharmaceutical industry. There is continued uncertainty about the implementation of ACA, including the potential for further amendments to the ACA and legal challenges to or efforts to repeal the ACA. We cannot be sure whether additional legislative changes will be enacted, or whether government regulations, guidance or interpretations will be changed, or what the impact of such changes would be on the marketing approvals, sales, pricing, or reimbursement of our drug candidates or products, if any, may be.
If the FDA approves any of our future products and reimbursement for those products is approved by any federal or state healthcare programs, then we will be subject to federal and state laws, such as the Federal False Claims Act, state false claims acts, the illegal remuneration provisions of the Social Security Act, and federal and state anti-kickback laws that govern financial and other arrangements among drug manufacturers and developers and the physicians and other practitioners or facilities that purchase or prescribe products. Among other things, these laws prohibit kickbacks, bribes and rebates, as well as other direct and indirect payments that are intended to induce the use or prescription of medical products or services payable by any federal or state healthcare program, and prohibit presenting a false or misleading claim for payment under a federal or state program. Possible sanctions for violation of any of these restrictions or prohibitions include loss of eligibility to participate in federal and state reimbursement programs and civil and criminal penalties. If we fail to comply, even inadvertently, with any of these requirements, we could be required to alter our operations, enter into corporate integrity, deferred prosecution or similar agreements with state or federal government agencies, and could become subject to significant civil and criminal penalties.
AVAILABLE INFORMATION
Our common stock is listed on The Nasdaq Capital Market and trades under the symbol “CWBR.” Our principal executive offices are located at 1455 Adams Drive, Suite 2050, Menlo Park, California 94025, and our telephone number is (650) 446-7888. The internet address of our corporate website is http://www.cohbar.com.
We file annual reports, quarterly reports, current reports, proxy statements and other information with the Securities and Exchange Commission (the “SEC”) under the Securities Exchange Act of 1934, as amended. Our filings with the SEC are available free of charge on the SEC’s website at www.sec.gov and on our website under the “Investors” tab as soon as reasonably practicable after we electronically file such material with, or furnish it to, the SEC.
The contents of our corporate website are not incorporated into, or otherwise to be regarded as part of, this Annual Report on Form 10-K.
16
Item 1A. Risk Factors
Summary of Risk Factors
An investment in our common stock involves various risks, and prospective investors are urged to carefully consider the matters discussed in the section titled “Risk Factors” prior to making an investment in our common stock. These risks include, but are not limited to, the following:
| ● | we are an early-stage biotechnology company and may never be able to successfully develop marketable products or generate any revenue. We have a limited relevant operating history upon which an evaluation of our performance and prospects can be made. There is no assurance that our future operations will result in profits. If we cannot generate sufficient revenues, we may suspend or cease operations; |
| ● | we have had a history of losses and no revenue; |
| ● | the outbreak of the novel strain of coronavirus, SARS-CoV-2, which causes COVID-19, and the ongoing COVID-19 pandemic, could adversely impact our business, including our clinical trials and preclinical studies; |
| ● | if we fail to demonstrate efficacy or safety in our research and clinical trials, our future business prospects, financial condition and operating results will be materially adversely affected; |
| ● | if any of our future clinical trials are delayed, suspended or terminated, we may be unable to develop our product candidates on a timely basis, which would adversely affect our ability to obtain regulatory approvals, increase our development costs and delay or prevent commercialization of any approved products; |
| ● | if we do not achieve our projected development goals in the time frames we announce and expect, the commercialization of our products may be delayed and, as a result, our stock price may decline; |
| ● | our future success depends on key members of our scientific team and our ability to attract, retain and motivate qualified personnel; |
| ● | we may seek to establish development and commercialization collaborations, and, if we are not able to establish them on commercially reasonable terms, we may have to alter our development and commercialization plans; |
| ● | we may not be successful in our efforts to identify or discover potential drug development candidates; |
| ● | our research and development plans will require substantial additional future funding which could impact our operational and financial condition. Without the required additional funds, we will likely cease operations; |
| ● | even if we are able to develop our potential drugs, we may not be able to obtain regulatory approval, or if approved, we may not be able to generate significant revenues or successfully commercialize our products, which will adversely affect our financial results and financial condition, and we will have to delay or terminate some or all of our research and development plans, which may force us to cease operations; |
| ● | if we do not maintain the support of qualified scientific collaborators, our revenue, growth and profitability will likely be limited, which would have a material adverse effect on our business; |
| ● | we expect to rely on third parties to conduct our clinical trials and some aspects of our research and preclinical testing. These third parties may not perform satisfactorily, including failing to meet deadlines for the completion of such trials, research or preclinical testing; |
| ● | we contract with third parties for the manufacture of our peptide materials for research and preclinical testing and expect to continue to do so for any future product candidate advanced to clinical trials and commercialization. This reliance on third parties increases the risk that we will not have sufficient quantities of our research peptide materials, product candidates or medicines, or that such supply will not be available to us at an acceptable cost, which could delay, prevent or impair our research, development or commercialization efforts; |
17
| ● | we may not be able to develop drug candidates, market or generate sales of our products to the extent anticipated. Our business may fail, and investors could lose all of their investment in our Company; |
| ● | interim and preliminary or topline data from our clinical trials that we announce or publish from time to time may change as more patient data become available and are subject to audit and verification procedures that could result in material changes in the final data; |
| ● | we expect to expand our drug development and regulatory capabilities, and as a result, we may encounter difficulties in managing our growth, which could disrupt our operations; and |
| ● | the use of any of our products in clinical trials may expose us to liability claims, which may cost us significant amounts of money to defend against or pay out, causing our business to suffer. |
CohBar operates in an environment that involves a number of risks and uncertainties. The risks and uncertainties described in this Annual Report on Form 10-K are not the only risks and uncertainties that we face. Additional risks and uncertainties that presently are not considered material or are not known to us, and therefore are not mentioned herein, may impair our business operations. If any of the risks described in this Annual Report on Form 10-K actually occur, our business, operating results and financial position could be adversely affected.
Risks Related to Our Financial Position and Need for Additional Capital
We have had a history of losses and no revenue.
We have generated substantial accumulated losses since our inception. We have not generated any revenues from our operations to date and do not expect to generate any revenue in the near future. As a result, our management expects the business to continue to experience negative cash flow for the foreseeable future. We can offer no assurance that we will ever operate profitably or that we will generate positive cash flow in the future.
Until we can generate significant revenues, if ever, we expect to satisfy our future cash needs through equity or debt financing. We will need to raise additional funds, and such funds may not be available on commercially acceptable terms, if at all. If we are unable to raise funds on acceptable terms, we may not be able to execute our business plan, take advantage of future opportunities, or respond to competitive pressures or unanticipated requirements. This may seriously harm our business, financial condition and results of operations. In the event we are not able to continue operations, investors will likely suffer a complete loss of their investments in our securities.
We are an early-stage biotechnology company and may never be able to successfully develop marketable products or generate any revenue. We have a limited relevant operating history upon which an evaluation of our performance and prospects can be made. There is no assurance that our future operations will result in profits. If we cannot generate sufficient revenues, we may suspend or cease operations.
We are an early-stage company. Our operations to date have been limited to organizing and staffing our Company, business planning, raising capital, identifying MDPs for further research, developing our intellectual property portfolio, performing research on identified MDPs and our novel analogs and progressing our most advanced drug candidate into and through clinical studies. We have not generated any revenues to date. All of our novel peptide analogs are in the concept, research or early clinical stages. Moreover, we cannot be certain that our research and development efforts will be successful or, if successful, that our novel peptide analogs will ever be approved by the FDA. Typically, it takes 10 to 12 years to develop one new medicine from the time it is discovered to when it is available for treating patients, and longer timeframes are not uncommon. Even if approved, our products may not generate commercial revenues. We have no relevant operating history upon which an evaluation of our performance and prospects can be made. We are subject to all of the business risks associated with a new enterprise, including, but not limited to, risks of unforeseen capital requirements, failure of potential drug candidates either in research, preclinical testing or in clinical trials, and failure to establish business relationships and competitive advantages against other companies. If we fail to become profitable, we may be forced to suspend or cease operations.
18
The outbreak of the novel strain of coronavirus, SARS-CoV-2, which causes COVID-19, and the ongoing COVID-19 pandemic, could adversely impact our business, including our clinical trials and preclinical studies.
Public health crises such as pandemics or similar outbreaks could adversely impact our business. In response to the global COVID-19 pandemic, we have modified our business practices by restricting nonessential travel, implementing a partial work from home policy for our employees and instituting new safety protocols for our lab to enable essential on-site work to continue. We continue to monitor the impact of COVID-19 on ongoing activities at our external research and development partner sites.
Timely enrollment in our clinical trials is dependent upon global clinical trial sites, which may be adversely affected by global health matters, such as pandemics. These and any additional delays in our clinical trials could increase our development costs, delay or prevent the availability of topline data expected to be available from the trial, delay our product development and regulatory submission process, result in the termination of the trial or make it difficult to raise additional capital.
As a result of the COVID-19 outbreak, or similar pandemics, we may experience disruptions that could severely impact our business, clinical trials and preclinical studies, including:
| ● | delays or difficulties in recruiting, enrolling and retaining patients in our clinical trials; |
| ● | delays or difficulties in clinical site initiation, including difficulties in recruiting clinical site investigators and clinical site staff; |
| ● | delays or disruptions in non-clinical experiments and investigational new drug application-enabling good laboratory practice standard toxicology studies due to unforeseen circumstances in the supply chain; |
| ● | increased rates of patients withdrawing from our clinical trials following enrollment as a result of contracting COVID-19, being forced to quarantine or not accepting home health visits; |
| ● | diversion of healthcare resources away from the conduct of clinical trials, including the diversion of hospitals serving as our clinical trial sites and hospital staff supporting the conduct of our clinical trials; |
| ● | interruption of key clinical trial activities, such as clinical trial site data monitoring, due to limitations on travel imposed or recommended by federal or state governments, employers and others or interruption of clinical trial subject visits and study procedures (particularly any procedures that may be deemed non-essential), which may impact the integrity of subject data and clinical study endpoints; |
| ● | interruption or delays in the operations of the FDA and comparable foreign regulatory agencies, which may impact approval timelines; |
| ● | limitations on employee resources that would otherwise be focused on the conduct of our preclinical studies and clinical trials, including because of sickness of employees or their families, the desire of employees to avoid contact with large groups of people, an increased reliance on working from home or mass transit disruptions; |
| ● | disruptions in the supply chain and the manufacture or shipment of both drug substance and finished drug product for our product candidates for preclinical testing and clinical trials; |
| ● | interruption of, or delays in receiving, supplies of our product candidates from our contract manufacturing organizations due to staffing shortages, production slowdowns or stoppages and disruptions in delivery systems; and |
| ● | reduced ability to engage with the medical, investor and partnering communities due to the cancellation of conferences scheduled throughout the year. |
19
In addition, the trading prices for our common stock and other biopharmaceutical companies have been highly volatile as a result of the COVID-19 pandemic and the resulting impact on economic activity. As COVID-19 transitions from a pandemic to an endemic disease, we are uncertain about its ongoing effect on both domestic and worldwide economic activity, which may continue to be unpredictable. As a result, we may face difficulties raising capital through sales of our common stock or other equity-linked securities, and any such sales may be on unfavorable terms to us and potentially dilutive to existing stockholders.
The extent to which the pandemic may impact our business, clinical trials and preclinical studies will depend on future developments, which are highly uncertain and cannot be predicted with confidence, such as the duration of the pandemic, the emergence of novel variants of SARS-CoV-2, the impact of vaccinations and vaccination rates, travel restrictions and actions to contain the virus or treat its impact, such as social distancing and quarantines or lock-downs in the United States and other countries, business closures or business disruptions and the effectiveness of actions taken in the United States and other countries to contain and treat the disease. For example, primarily due to COVID-19 related restrictions and disruption, we have experienced delays in shipping raw materials to our partners in China, which has delayed certain of our investigational new drug application-enabling activities.
If we fail to demonstrate efficacy or safety in our research and clinical trials, our future business prospects, financial condition and operating results will be materially adversely affected.
The success of our research and development efforts will greatly depend on our ability to demonstrate efficacy of our novel peptide analogs in non-clinical studies, as well as in clinical trials. Non-clinical studies involve testing potential drug candidates in appropriate non-human disease models to demonstrate efficacy and safety. Regulatory agencies evaluate these data carefully before they will approve clinical testing in humans. If certain non-clinical data reveals potential safety issues or the results are inconsistent with an expectation of the potential drug’s efficacy in humans, the program may be discontinued or the regulatory agencies may require additional testing before allowing human clinical trials. This additional testing will increase program expenses and extend timelines. We may decide to suspend further testing on our potential drugs if, in the judgment of our management and advisors, the non-clinical test results do not support further development.
Moreover, success in research, preclinical testing and early clinical trials does not ensure that later clinical trials will be successful, and we cannot be sure that the results of later clinical trials will replicate the results of prior clinical trials and non-clinical testing. The clinical trial process may fail to demonstrate that our potential drug candidates are safe for humans and effective for indicated uses. This failure would cause us to abandon a drug candidate and may delay development of other potential drug candidates. Any delay in, or termination of, our non-clinical testing or clinical trials will delay the filing of an investigational new drug application and new drug application with the FDA or the equivalent applications with pharmaceutical regulatory authorities outside the United States and, ultimately, our ability to commercialize our potential drugs and generate product revenues. In addition, our Phase 1a/1b trial of CB4211, our most advanced drug candidate, involved, and we expect that our other early clinical trials will involve, small patient populations. Because of these small sample sizes, the results of these early clinical trials, including the topline data from our CB4211 Phase 1a/1b trial, may not be indicative of future results.
Risks Related to Discovery, Development and Commercialization
If any of our future clinical trials are delayed, suspended or terminated, we may be unable to develop our product candidates on a timely basis, which would adversely affect our ability to obtain regulatory approvals, increase our development costs and delay or prevent commercialization of any approved products.
We cannot predict whether we will encounter problems with any of our planned or future clinical trials that will cause regulatory agencies, institutional review boards, or us to suspend or delay a trial. For example, in November 2018, the Company announced the temporary suspension of the Phase 1a/1b clinical trial for CB4211 in order to address injection site reactions, and we resumed the trial in June 2019. In November 2019, we announced the completion of the Phase 1a portion of the clinical trial and the commencement of the recruiting phase of the final Phase 1b stage of the study. However, in March 2020, we announced a delay in the completion of this trial due to a pause by some of our clinical research organization partners in all of their activities related to the study in response to developments relating to the COVID-19 pandemic. While we announced the resumption of our Phase 1b study in July 2020, our clinical activities could be delayed again in the future. Additionally, the FDA’s review of any prior or future submissions related to completed, ongoing, or planned clinical trials of our product candidates, or future information requests from the FDA could result in the delay or suspension of any ongoing or planned clinical trials to address any concerns.
20
Clinical trials and clinical data collection protocols can be delayed for a variety of reasons, including:
| ● | unanticipated consequences of the formulation of the product candidate requiring us to pause the trial to investigate alternative formulations; |
| ● | the occurrence of unacceptable drug-related side effects or adverse events experienced by participants in our clinical trials; |
| ● | discussions with the FDA regarding the scope or design of our clinical trials and clinical data collection protocols; |
| ● | delays or the inability to obtain required approvals from institutional review boards or other responsible entities at clinical sites selected for participation in our existing or future clinical trials; |
| ● | adverse findings in clinical or nonclinical studies related to the safety of our product candidates in humans; |
| ● | the amendment of clinical trial or data collection protocols to reflect changes in regulatory requirements and guidance or other reasons, as well as subsequent re-examination of amendments of clinical trial or data collection protocols by institutional review boards or other responsible bodies; and |
| ● | the need to repeat or conduct additional clinical trials as a result of inconclusive or negative results, failure to replicate positive early clinical data in subsequent clinical trials, failure to deliver an efficacious dose of a product candidate, poorly executed testing, a failure of a clinical site to adhere to the clinical protocol, an unacceptable study design or other problems. |
In addition, a clinical trial or development program may be suspended or terminated by us, institutional review boards, the FDA or other responsible bodies due to a number of factors, including:
| ● | failure to conduct the clinical trial in accordance with regulatory requirements or our clinical protocols; |
| ● | inspection of the clinical trial operations or trial sites by the FDA or other regulatory authorities resulting in the imposition of a clinical hold; |
| ● | inability to resume a suspended trial in a timely manner, which we cannot predict with certainty, if at all; |
| ● | unforeseen safety issues or any determination that a trial presents unacceptable health risks; |
| ● | inability to deliver an efficacious dose of a product candidate; and |
| ● | lack of adequate funding to continue the clinical trial. |
If the results of our clinical trials are not available when we expect or if we encounter any delay in the analysis of data from our clinical trials, we may be unable to conduct additional clinical trials on the schedule we anticipate. Many of the factors that cause, or lead to, a delay in the commencement or completion of clinical trials may also ultimately lead to the denial of regulatory approval of a product candidate. Any delays in completing a clinical trial could increase our development costs, delay or prevent the availability of topline data expected to be available from the trial, delay our product development and regulatory submission process or make it difficult to raise additional capital.
If we do not achieve our projected development goals in the time frames we announce and expect, the commercialization of our products may be delayed and, as a result, our stock price may decline.
From time to time, we estimate the timing of the anticipated accomplishment of various scientific, clinical, regulatory and other product development goals, which we sometimes refer to as milestones. These milestones may include the commencement or completion of scientific studies and clinical trials and the submission of regulatory filings. From time to time, we may publicly announce the expected timing of some of these milestones. All of these milestones are and will be based on numerous assumptions, including timely performance by our contract research organizations (“CROs”) and other vendors, positive clinical and preclinical results, the addition of a corporate partner for our CB4211 program, and sufficient funding from partnering and general fundraising. The actual timing of these milestones can vary dramatically compared to our estimates, in some cases for reasons beyond our control. If we do not meet these milestones as publicly announced, or at all, our revenue may be lower than expected, the commercialization of our products may be delayed or never achieved and, as a result, our stock price may decline.
21
Our future success depends on key members of our management and scientific teams and our ability to attract, retain and motivate qualified personnel.
Recruiting and retaining qualified senior management and scientific, clinical, and operations management and personnel will be critical to our success. We may not be able to attract and retain these personnel on acceptable terms given the competition among numerous pharmaceutical and biopharmaceutical companies for similar personnel. We also experience competition for the hiring of scientific and clinical personnel from universities and research institutions.
We are highly dependent on our key management and scientific teams, including our Chief Executive Officer and Chief Financial Officer who are employed “at will,” meaning they may terminate the employment relationship at any time. We do not maintain “key person” insurance for any of the key members of our team. The loss of the services of any of these persons could impede the achievement of our research, development and commercialization objectives. We have in the past and may in the future continue to experience changes in our executive management team resulting from the departure of executives or subsequent hiring of new executives, which may be disruptive to our business. For example, Kenneth Cundy resigned from his role of Chief Scientific Officer effective March 31, 2022. We anticipate that we will experience a transitional period as other members of the team assume Dr. Cundy’s responsibilities, and such transition may have a disruptive impact on our ability to implement our business strategy and could have a material adverse effect on our business. Any changes in business strategies can create uncertainty, may negatively impact our ability to execute our business strategy quickly and effectively and may ultimately be unsuccessful. The impact of hiring new executives may not be immediately realized.
We rely on consultants and advisors from time to time, including drug discovery and development advisors, to assist us in formulating our research and development strategy. Agreements with these advisors typically may be terminated by either party, for any reason, on relatively short notice. In addition, our consultants and advisors, including our founders, may be employed by employers other than us and may have commitments under consulting or advisory contracts with other entities that may limit their availability to us.
We may seek to establish development and commercialization collaborations, and, if we are not able to establish them on commercially reasonable terms, we may have to alter our development and commercialization plans.
Our potential drug development programs and the potential commercialization of our drug candidates will require substantial additional cash to fund expenses. We may decide to collaborate with biopharmaceutical or biotechnology companies in connection with the development or commercialization of our potential drug candidates. For example, we intend to partner CB4211 before moving this program forward into further clinical trials. There is no guarantee that we will be able to establish a partnership for the CB4211 program on favorable terms, if at all. If we are unable to establish such a partnership, our CB4211 program may be delayed or terminated, which may cause our stock price to decline or otherwise result in adverse effects on our business.
We face significant competition in seeking appropriate collaborators. Whether we reach a definitive collaboration agreement will depend, among other things, upon our assessment of the collaborator’s resources and expertise, the terms and conditions of the proposed collaboration and the proposed collaborator’s evaluation of a number of factors. Those factors may include the design or results of clinical trials, the likelihood of approval by the FDA or similar regulatory authorities outside the United States, the potential market for the subject product candidate, the costs and complexities of manufacturing and delivering such product candidate to patients, the potential reimbursement rates for such product candidates, the potential of competing products, the existence of uncertainty with respect to our ownership of technology, which can exist if there is a challenge to such ownership without regard to the merits of the challenge, and industry and market conditions generally. The collaborator may also consider alternative product candidates or technologies for similar disease indications on which to collaborate, and whether such alternative collaboration project could be more attractive than one with us for our product candidate.
There are a limited number of large biopharmaceutical companies with whom we could potentially collaborate, and collaborations are complex and time-consuming to negotiate and document. We may not be able to negotiate collaborations on a timely basis, on acceptable terms or at all. If we are unable to do so, we may have to curtail the development of the product candidate for which we are seeking to collaborate, reduce or delay its development program or one or more of our other development programs, delay its potential commercialization or reduce the scope of any sales or marketing activities, or increase our expenditures and undertake development or commercialization activities at our own expense. If we elect to increase our expenditures to fund development or commercialization activities on our own, we may need to obtain additional capital, which may not be available to us on acceptable terms or at all. If we do not have sufficient funds, we may not be able to further develop our product candidates or bring them to market and generate product revenue.
22
We may not be successful in our efforts to identify or discover potential drug development candidates.
A key element of our strategy is to identify and test MDPs and novel analogs that play a role in cellular processes underlying our targeted disease indications. A significant portion of the research that we are conducting involves emerging scientific knowledge and drug discovery methods. Our drug discovery efforts may not be successful in identifying novel peptide analogs that are useful in treating disease. Our research programs may initially show promise in identifying potential drug development candidates, yet fail to yield candidates for preclinical and clinical development for a number of reasons, including:
| ● | the research methodology used may not be successful in identifying appropriate potential drug development candidates; |
| ● | we may not be able to identify the mechanism of action for potential drug candidates, which may make it more difficult to develop and commercialize such drug candidates due to the potential desire of the FDA and other regulatory bodies, potential partners, physicians and patients to understand such mechanism of action; or |
| ● | potential drug development candidates may, on further study, be shown not to be effective in humans, or to have unacceptable toxicities, harmful side effects, properties that make them difficult or impossible to formulate in a commercial fashion, or other characteristics that indicate that they are unlikely to be medicines that will receive marketing approval and achieve market acceptance. |
Research programs to identify new product candidates require substantial technical, financial and human resources. We may choose to focus our efforts and resources on a potential product candidate that ultimately proves to be unsuccessful. As a result, we may forego or delay pursuit of opportunities with other product candidates or for other disease indications that later prove to have greater commercial potential. Our resource allocation decisions may cause us to fail to timely capitalize on viable commercial products or profitable market opportunities. If we are unable to progress our most advanced drug candidate through clinical development or identify other novel peptide analogs that are suitable for preclinical and clinical development, we will not be able to generate product revenues in future periods, which likely would result in significant harm to our financial position and negatively affect our ability to continue our operations.
Our research and development plans will require substantial additional future funding which could impact our operational and financial condition. Without the required additional funds, we will likely cease operations.
It will take several years before we are able to develop potentially marketable products, if at all. Our research and development plans will require substantial additional capital to:
| ● | conduct research, preclinical testing and human studies; |
| ● | manufacture any future drug development candidate or product at pilot and commercial scale; and |
| ● | establish and develop quality control, regulatory, and administrative capabilities to support these programs. |
Our future operating and capital needs will depend on many factors, including:
| ● | the pace of scientific progress in our research programs and the magnitude of these programs; |
| ● | the scope and results of preclinical testing and human studies; |
| ● | the time and costs involved in obtaining regulatory approvals; |
| ● | the time and costs involved in preparing, filing, prosecuting, securing, maintaining and enforcing intellectual property rights; |
| ● | competing technological and market developments; |
| ● | our ability to establish additional collaborations; |
23
| ● | changes in any future collaborations; |
| ● | the cost of manufacturing our drug products; and |
| ● | the cost and effectiveness of efforts to commercialize and market our products. |
We base our outlook regarding the need for funds on many uncertain variables. Such uncertainties include the success of our research and development initiatives, regulatory approvals, the timing of events outside our direct control such as negotiations with potential strategic partners, and other factors. Any of these uncertain events can significantly change our cash requirements as they determine such one-time events as the receipt or payment of major milestones and other payments.
Additional funds will be required to support our operations, and if we are unable to obtain them on favorable terms, we may be required to cease or reduce further research and development of our drug product programs, sell or abandon some or all of our intellectual property, merge with another entity or cease operations.
Even if we are able to develop our potential drugs, we may not be able to obtain regulatory approval, or if approved, we may not be able to generate significant revenues or successfully commercialize our products, which will adversely affect our financial results and financial condition, and we will have to delay or terminate some or all of our research and development plans, which may force us to cease operations.
All our potential drug candidates will require extensive additional research and development, including preclinical testing and clinical trials, as well as regulatory approvals, before we can market them. We cannot predict if or when any potential drug candidate we intend to develop will be approved for marketing. There are many reasons that we may fail in our efforts to develop our potential drug candidates. These include:
| ● | the possibility that preclinical testing or clinical trials may show that our potential drugs are ineffective and/or cause harmful side effects or toxicities; |
| ● | we may not be able to develop commercially viable formulations for our potential drug candidates; |
| ● | our potential drugs may prove to be too expensive to manufacture or administer to patients; |
| ● | our potential drugs may fail to receive necessary regulatory approvals from the FDA or foreign regulatory authorities in a timely manner, or at all; |
| ● | even if our potential drugs are approved, we may not be able to produce them in commercial quantities or at reasonable costs; |
| ● | even if our potential drugs are approved, they may not achieve commercial acceptance; |
| ● | even if our potential drugs are approved and commercially launched, they may not receive desirable payor reimbursement and formulary access; |
| ● | regulatory or governmental authorities may apply restrictions to any of our potential drugs, which could adversely affect their commercial success; and |
| ● | the proprietary rights of other parties may prevent us or our potential collaborative partners from marketing our potential drugs. |
If we fail to develop our potential drug candidates, our financial results and financial condition will be adversely affected, we will have to delay or terminate some or all of our research and development plans and may be forced to cease operations.
24
Risks Related to Our Reliance on Third Parties
If we do not maintain the support of qualified scientific collaborators, our revenue, growth and profitability will likely be limited, which would have a material adverse effect on our business.
We will need to maintain our existing relationships with leading scientists and/or establish new relationships with scientific collaborators. We believe that such relationships are pivotal to establishing products using our technologies as a standard of care for various disease indications. There is no assurance that our founders, scientific advisors or research partners will continue to work with us or that we will be able to attract additional research partners. If we are not able to establish scientific relationships to assist in our research and development, we may not be able to successfully develop our potential drug candidates. If this happens, our business will be adversely affected.
We expect to rely on third parties to conduct our clinical trials and some aspects of our research and preclinical testing. These third parties may not perform satisfactorily, including failing to meet deadlines for the completion of such trials, research or preclinical testing.
We currently rely on third parties to conduct some aspects of our research and expect to continue to rely on third parties to conduct additional aspects of our research and preclinical testing, as well as any future clinical trials. Any of these third parties may terminate their engagements with us at any time. If we need to enter into alternative arrangements, it would delay our product research and development activities.
Our reliance on these third parties for research and development activities will reduce our control over these activities but will not relieve us of our responsibilities. For example, we will remain responsible for ensuring that each of our clinical trials is conducted in accordance with the general investigational plan and protocols for the trial. Moreover, the FDA requires us to comply with standards, commonly referred to as Good Clinical Practices, for conducting, recording and reporting the results of clinical trials to assure that data and reported results are credible and accurate and that the rights, integrity and confidentiality of trial participants are protected. We also are required to register ongoing clinical trials and post the results of completed clinical trials on a government-sponsored database, ClinicalTrials.gov, within certain timeframes. Failure to do so can result in fines, adverse publicity and civil and criminal sanctions.
Furthermore, these third parties may also have relationships with other entities, some of which may be our competitors. If these third parties do not successfully carry out their contractual duties, meet expected deadlines or conduct our clinical trials in accordance with regulatory requirements or our stated protocols, we will not be able to obtain, or may be delayed in obtaining, marketing approvals for our drug candidates and will not be able to, or may be delayed in our efforts to, successfully commercialize our medicines. For example, we experienced delays in receiving the data from our third-party CRO conducting our CB4211 Phase 1b study, which delayed our analysis and release of topline data.
We currently rely, and expect to continue to rely, on other third parties to store and distribute drug supplies for our clinical trials. Any performance failure on the part of our distributors could delay clinical development or marketing approval of our drug candidates or commercialization of our products, producing additional losses and depriving us of potential product revenue.
We contract with third parties for the manufacture of our peptide materials for research and preclinical testing and expect to continue to do so for any future product candidate advanced to clinical trials and commercialization. This reliance on third parties increases the risk that we will not have sufficient quantities of our research peptide materials, product candidates or medicines, or that such supply will not be available to us at an acceptable cost, which could delay, prevent or impair our research, development or commercialization efforts.
We do not have manufacturing facilities adequate to produce our research peptide materials or supplies of any future product candidate. We currently rely, and expect to continue to rely, on third-party manufacturers for the manufacture of our peptide materials, our current and any future product candidates for preclinical and clinical testing, and for commercial supply of any of these product candidates for which we or future collaborators obtain marketing approval. We do not have long term supply agreements with any third-party manufacturers, and we purchase our research peptides on a purchase order basis.
25
We may be unable to establish any agreements with third-party manufacturers or to do so on acceptable terms. Even if we are able to establish agreements with third-party manufacturers, reliance on third-party manufacturers entails additional risks, including:
| ● | reliance on the third party for producing the peptide materials or product candidates according to the detailed specifications; |
| ● | reliance on the third party for regulatory compliance and quality assurance; |
| ● | the possible breach of the manufacturing agreement by the third party; |
| ● | the possible termination or nonrenewal of the agreement by the third party at a time that is costly or inconvenient for us; and |
| ● | reliance on the third party for regulatory compliance, quality assurance, and safety and pharmacovigilance reporting. |
Third-party manufacturers may not be able to comply with cGMP as enforced by the FDA, or regulations or similar regulatory requirements outside the United States. Our failure, or the failure of our third-party manufacturers, to comply with applicable regulations could result in us being subject to sanctions, including fines, injunctions, civil penalties, delays, suspension or withdrawal of approvals, license revocation, seizures or recalls of product candidates or medicines, operating restrictions and criminal prosecutions, any of which could significantly and adversely affect supplies of our medicines and harm our business and results of operations.
Any drug candidate that we may develop may compete with other drug candidates and products for access to manufacturing facilities. There are a limited number of manufacturers that operate under cGMP regulations and that might be capable of manufacturing for us.
Our current and anticipated future dependence upon others for the manufacture of our investigational materials or future product candidates or medicines may adversely affect our future profit margins and our ability to commercialize any medicines that receive marketing approval on a timely and competitive basis.
Risks Related to Product Development and Regulatory Approval
Even if we are successful in developing drug candidates, we may not be able to market or generate sales of our products to the extent anticipated. Our business may fail, and investors could lose all of their investment in our Company.
Assuming that we are successful in developing our potential drug candidates and receiving regulatory clearances to market our potential products, our ability to successfully penetrate the market and generate sales of those products may be limited by a number of factors, including the following:
| ● | if our competitors receive regulatory approvals for and begin marketing similar products in the United States, the European Union (“EU”), Japan and other territories before we do, greater awareness of their products as compared to ours will cause our competitive position to suffer; |
| ● | information from our competitors or the academic community indicating that current products or new products are more effective or offer compelling other benefits than our future products could impede our market penetration or decrease our future market share; and |
| ● | the pricing and reimbursement environment for our future products, as well as pricing and reimbursement decisions by our competitors and by payers, may have an effect on our revenues. |
If any of these occur, our business could be adversely affected.
26
Interim and preliminary or topline data from our clinical trials that we announce or publish from time to time may change as more patient data become available and are subject to audit and verification procedures that could result in material changes in the final data.
From time to time, we may publish interim topline or preliminary data from our clinical trials. Interim data from clinical trials that we may complete are subject to the risk that one or more of the clinical outcomes may materially change as patient enrollment continues and more patient data become available. Preliminary or topline data also remain subject to audit and verification procedures that may result in the final data being materially different from the preliminary or topline data we previously published. As a result, interim and preliminary data should be viewed with caution until the final data are available. Adverse differences between interim or preliminary or topline data and final data could significantly harm our reputation and business prospects.
Any product candidate we are able to develop and commercialize would compete in the marketplace with existing therapies and new therapies that may become available in the future. These competitive therapies may be more effective, safer, less costly, more easily administered or offer other advantages over any product we seek to market.
Although there are no currently approved therapies for the treatment of NASH, there are numerous therapies in development, including those in clinical trials that are more advanced than ours. Additionally, there are numerous therapies currently marketed to treat IPF, diabetes, cancer, and other diseases for which our potential product candidates may be indicated. These therapies are varied in their design, therapeutic application and mechanism of action and may provide significant competition for any of our product candidates for which we obtain market approval. New products may also become available that provide efficacy, safety, convenience and other benefits that are not provided by currently marketed therapies. As a result, they may provide significant competition for any of our product candidates for which we obtain market approval. Our commercial opportunity could be reduced or eliminated if our competitors develop and commercialize products that are safer, more effective, have fewer or less severe side effects, are more conveniently administered or stored or are less expensive than any products that we may develop. Our competitors also may obtain FDA or other regulatory approval for their products more rapidly than we may obtain approval for ours, which could result in our competitors establishing a strong market position before we are able to enter the market. In addition, our ability to compete may be affected in many cases by insurers’ or other third-party payers’ reimbursement polices seeking to encourage the use of existing products which are generic or are otherwise less expensive to provide.
We expect to expand our drug development and regulatory capabilities, and as a result, we may encounter difficulties in managing our growth, which could disrupt our operations.
We expect to experience significant growth in the scope of our operations, particularly in the areas of drug development and commercialization and regulatory affairs. To manage our anticipated future growth, we must continue to implement and improve our managerial, operational and financial systems, expand our facilities and continue to recruit and train additional qualified personnel, which we may not be able to attract. We expect that if our drug candidates continue to progress into and in development, we may require significant additional investment in personnel, management systems and resources, particularly in the build out of our clinical and commercial capabilities. Over the next several years, we may experience significant growth in the number of our employees and the scope of our operations, particularly in the areas of drug development, regulatory affairs and sales and marketing. Due to our limited financial resources and our limited operating history, we may not be able to effectively manage the expected expansion of our operations. The physical expansion of our operations may lead to significant costs and may divert our management and business development resources. Any inability to manage growth could delay the execution of our business plans or disrupt our operations.
The use of any of our products in clinical trials, and the results of those trials, may expose us to liability claims, which may cost us significant amounts of money to defend against or pay out, causing our business to suffer.
The nature of our business exposes us to potential liability risks inherent in the testing, manufacturing and marketing of our products. If any of our drug candidates are used in clinical trials, or if any of our drug candidates become marketed products, they could potentially harm people or allegedly harm people, possibly subjecting us to costly and damaging product liability claims. Some of the patients who participate in clinical trials are already ill when they enter a trial or may intentionally or unintentionally fail to meet the exclusion criteria. The waivers we obtain may not be enforceable and may not protect us from liability or the costs of product liability litigation. Although we obtained product liability insurance, which we believe is adequate, we are subject to the risk that our insurance will not be sufficient to cover claims. We anticipate that we will need to increase our insurance coverage if we successfully commercialize any product candidate. The insurance costs along with the defense or payment of liabilities above the amount of coverage could cost us significant amounts of money and management distraction from other elements of the business, decrease demand for any product candidates that we may develop, injure our reputation and attract significant negative media attention, and lead to the withdrawal of clinical trial participants, causing our business to suffer. We may not be able to maintain insurance coverage at a reasonable cost or in an amount adequate to satisfy any liability that may arise.
27
Compliance with laws and regulations pertaining to the privacy and security of health information may be time consuming, difficult and costly, particularly in light of increased focus on privacy issues in countries around the world, including the United States and the EU.
We are subject to various domestic and international privacy and security regulations. The confidentiality, collection, use and disclosure of personal data, including clinical trial patient-specific information, are subject to governmental regulation generally in the country that the personal data were collected or used. In the United States, we are subject, or expect to be subject, to various state and federal privacy and data security regulations, including but not limited to the Health Insurance Portability and Accountability Act of 1996 (“HIPAA”), as amended by the Health Information Technology for Economic and Clinical Health Act of 2009. HIPAA mandates, among other things, the adoption of uniform standards for the electronic exchange of information in common health care transactions, as well as standards relating to the privacy and security of individually identifiable health information, which require the adoption of administrative, physical and technical safeguards to protect such information. In the EU, personal data includes any information that relates to an identified or identifiable natural person with health information carrying additional obligations, including obtaining the explicit consent from the individual for collection, use or disclosure of the information. In addition, the protection of and cross-border transfers of such data out of the EU has become more stringent with the EU’s General Data Protection Regulation which came into effect in May 2018. Furthermore, the legislative and regulatory landscape for privacy and data protection continues to evolve, and there has been an increasing amount of focus on privacy and data protection issues. The United States and the EU and its member states continue to issue new privacy and data protection rules and regulations that relate to personal data and health information. Compliance with these laws may be time consuming, difficult and costly. If we fail to comply with applicable laws, regulations or duties relating to the use, privacy or security of personal data, we could be subject to the imposition of significant civil and criminal penalties, be forced to alter our business practices and suffer reputational harm.
We may not be able to obtain agreement with regulatory authorities regarding an acceptable development plan for our product candidates, the outcome of our clinical trials may not be favorable or, even if favorable, regulatory authorities may not find the results of our clinical trials to be sufficient for marketing approval.
In the United States, the FDA generally requires two adequate and well-controlled pivotal clinical trials to approve a new drug application (“NDA”). Furthermore, for full approval of an NDA, the FDA requires a demonstration of efficacy based on a clinical benefit endpoint. The FDA may grant accelerated approval based on a surrogate endpoint reasonably likely to predict clinical benefit. Even though our pivotal clinical trials for a specific indication may achieve their primary endpoints and may be reasonably believed by us to be likely to predict clinical benefit, the FDA may not accept the results of such trials or approve our product candidates on an accelerated basis, or at all. It is also possible that the FDA may refuse to accept for filing and review any regulatory application we submit for regulatory approval in the United States. Even if our regulatory application is accepted for review, there may be delays in the FDA’s review process and the FDA may determine that such regulatory application does not contain adequate clinical or other data or support the approval of our product candidate. In such a case, the FDA may issue a complete response letter that may require that we conduct and/or complete additional clinical trials and preclinical studies or provide additional information or data before it will reconsider an application for approval. Any such requirements may be substantial, expensive and time-consuming, and there is no guarantee that we will continue to pursue such application or that the FDA will ultimately decide that any such application supports the approval of our product candidate. Furthermore, the FDA may also refer any regulatory application to an advisory committee for review and recommendation as to whether, and under what conditions, the application should be approved. While the FDA is not bound by the recommendation of an advisory committee, it considers such recommendations carefully when making decisions. Delay or failure to obtain, or unexpected costs in obtaining, the regulatory approval necessary to bring a potential product to market could decrease our ability to generate sufficient revenue to maintain our business.
The regulatory approval process is lengthy, expensive and uncertain, and we may be unable to obtain regulatory approval for our product candidates under applicable regulatory requirements. The denial or delay of any such approval would delay commercialization of our product candidates and adversely impact our ability to generate revenue, our business and our results of operations.
The development, research, testing, manufacturing, labeling, approval, selling, import, export, marketing, promotion and distribution of drug products are subject to extensive and evolving regulation by federal, state and local governmental authorities in the United States, principally the FDA, and by foreign regulatory authorities, which regulations differ from country to country. Neither we nor any future collaborator is permitted to market any of our product candidates in the United States until we receive regulatory approval of an NDA from the FDA.
28
Obtaining regulatory approval of an NDA can be a lengthy, expensive and uncertain process. Prior to obtaining approval to commercialize our product candidate in the United States or abroad, we or our collaborators must demonstrate with substantial evidence from well-controlled clinical trials, and to the satisfaction of the FDA or other foreign regulatory authorities, that such product candidates are safe and effective for their intended uses. The number of nonclinical studies and clinical trials that will be required for regulatory approval varies depending on the product candidate, the disease or condition that the product candidate is designed to address, and the regulations applicable to any particular product candidate.
Results from nonclinical studies and clinical trials can be interpreted in different ways. Even if we believe the nonclinical or clinical data for our product candidates are promising, such data may not be sufficient to support approval by the FDA and other regulatory authorities. Administering product candidates to humans may produce undesirable side effects, which could interrupt, delay or halt clinical trials and result in the FDA or other regulatory authorities denying approval of a product candidate for any or all indications. The FDA may also require us to conduct additional studies or trials for our product candidates either prior to or post-approval, such as additional clinical pharmacology studies or safety or efficacy studies or trials, or it may object to elements of our clinical development program such as the primary endpoints or the number of subjects in our clinical trials.
The FDA or any foreign regulatory bodies can delay, limit or deny approval of our product candidates or require us to conduct additional nonclinical or clinical testing or abandon a program for many reasons, including:
| ● | the FDA or the applicable foreign regulatory authority’s disagreement with the design or implementation of our clinical trials; |
| ● | negative or ambiguous results from our clinical trials or results that may not meet the level of statistical significance required by the FDA or comparable foreign regulatory authorities for approval; |
| ● | serious and unexpected drug-related side effects experienced by participants in our clinical trials; |
| ● | our inability to demonstrate to the satisfaction of the FDA or the applicable foreign regulatory authority that our product candidates are safe and effective for the proposed indication; |
| ● | the FDA’s or the applicable foreign regulatory authority’s disagreement with the interpretation of data from nonclinical studies or clinical trials; |
| ● | our inability to demonstrate the clinical and other benefits of our product candidates outweigh any safety or other perceived risks; |
| ● | the FDA’s or the applicable foreign regulatory authority’s requirement for additional nonclinical studies or clinical trials; |
| ● | the FDA’s or the applicable foreign regulatory authority’s disagreement regarding the formulation, labeling and/or the specifications of our product candidates; |
| ● | the FDA’s or the applicable foreign regulatory authority’s failure to approve the manufacturing processes or facilities of third-party manufacturers with which we contract; |
| ● | the potential for approval policies or regulations of the FDA or the applicable foreign regulatory authorities to significantly change in a manner rendering our clinical data insufficient for approval; or |
| ● | the FDA or the applicable foreign regulatory authority’s disagreement with the sufficiency of the clinical, non-clinical and/or quality data in the NDA or comparable marketing authorization application. |
Of the large number of drugs in development, only a small percentage successfully complete the FDA or other regulatory approval processes and are commercialized. The lengthy development and approval process as well as the unpredictability of future clinical trial results may result in our failing to obtain regulatory approval to market our product candidates, which would significantly harm our business, financial condition, results of operations and prospects.
29
Any product candidate for which we obtain marketing approval will be subject to extensive post-marketing regulatory requirements and could be subject to post-marketing restrictions or withdrawal from the market, and we may be subject to penalties if we fail to comply with regulatory requirements or if we experience unanticipated problems with our product candidates, when and if any of them are approved.
Our product candidates and the activities associated with their development and potential commercialization, including their testing, manufacturing, recordkeeping, labeling, storage, approval, advertising, promotion, sale and distribution, are subject to comprehensive regulation by the FDA and other U.S. and international regulatory authorities. These requirements include submissions of safety and other post-marketing information and reports, registration and listing requirements, requirements relating to manufacturing, including current cGMPs, quality control, quality assurance and corresponding maintenance of records and documents, including periodic inspections by the FDA and other regulatory authorities and requirements regarding the distribution of samples to providers and recordkeeping.
The FDA may also impose requirements for costly post-marketing studies or clinical trials and surveillance to monitor the safety or efficacy of any approved product. The FDA closely regulates the post-approval marketing and promotion of drugs and biologics to ensure drugs and biologics are marketed only for the approved disease indications and in accordance with the provisions of the approved labeling. The FDA imposes stringent restrictions on manufacturers’ communications regarding use of their products. If we promote our product candidates in a manner inconsistent with FDA-approved labeling or otherwise not in compliance with FDA regulations, we may be subject to enforcement action. Violations of the FDCA relating to the promotion of prescription drugs may lead to investigations alleging violations of federal and state healthcare fraud and abuse laws, as well as state consumer protection laws and similar laws in international jurisdictions.
In addition, later discovery of previously unknown adverse events or other problems with our product candidates, manufacturers or manufacturing processes, or failure to comply with regulatory requirements, may yield various results, including:
| ● | restrictions on such product candidates, manufacturers or manufacturing processes; |
| ● | restrictions on the labeling or marketing of a product; |
| ● | restrictions on product distribution or use; |
| ● | requirements to conduct post-marketing studies or clinical trials; |
| ● | warning or untitled letters; |
| ● | withdrawal of any approved product from the market; |
| ● | refusal to approve pending applications or supplements to approved applications that we submit; |
| ● | recall of product candidates; |
| ● | restrictions on product distribution or use; |
| ● | fines, restitution or disgorgement of profits or revenues; |
| ● | suspension or withdrawal of marketing approvals; |
| ● | refusal to permit the import or export of our product candidates; |
| ● | product seizure; and |
| ● | injunctions or the imposition of civil or criminal penalties. |
Non-compliance with European requirements regarding safety monitoring or pharmacovigilance, and with requirements related to the development of products for the pediatric population, can also result in significant financial penalties. Similarly, failure to comply with the EU’s requirements regarding the protection of personal information can also lead to significant penalties and sanctions.
30
The patent positions of biopharmaceutical products are complex and uncertain, and we may not be able to protect our patented or other intellectual property. If we cannot protect this property, we may be prevented from using it, or our competitors may use it, and our business could suffer significant harm. Also, the time and money we spend on acquiring and enforcing patents and other intellectual property will reduce the time and money we have available for our research and development, possibly resulting in a slow down or cessation of our research and development.
We own or exclusively license patents and patent applications related to our MDPs and potential drug candidates comprised of novel analogs and we anticipate continuing to develop our intellectual property portfolio. However, neither patents nor patent applications ensure the protection of our intellectual property for a number of reasons, including the following:
| ● | The United States Supreme Court rendered a decision in Molecular Pathology vs. Myriad Genetics, Inc., 133 S.Ct. 2107 (2013) (“Myriad”), in which the court held that naturally occurring DNA segments are products of nature and not patentable as compositions of matter. On March 4, 2014, the USPTO issued guidelines for examination of such claims that, among other things, extended the Myriad decision to any natural product. Since MDPs are natural products isolated from cells, the USPTO guidelines may affect allowability of some of our patent claims (pertaining to natural MDP sequences) that are filed in the USPTO but are not yet issued. Further, while the USPTO guidelines are not binding on the courts, it is likely that as the law of subject matter eligibility continues to develop, Myriad will be extended to natural products other than DNA. Thus, our issued U.S. patent claims directed to MDPs as compositions of matter may be vulnerable to challenge by competitors who seek to have our claims rendered invalid. While Myriad and the USPTO guidelines described above will affect our patents only in the United States, there is no certainty that similar laws or regulations will not be adopted in other jurisdictions. |
| ● | Competitors may interfere with our patenting process in a variety of ways. Competitors may claim that they invented the claimed invention prior to us. Competitors may also claim that we are infringing their patents and restrict our freedom to operate. Competitors may also contest our patents and patent applications, if issued, by showing in various patent offices that, among other reasons, the patented subject matter was not original, was not novel or was obvious. In litigation, a competitor could claim that our patents and patent applications are not valid or enforceable for a number of reasons. If a court agrees, we would lose some or all of our patent protection. |
| ● | As a company, we have no meaningful experience with competitors interfering with our patents or patent applications. In order to enforce our intellectual property, we may need to file a lawsuit against a competitor. Enforcing our intellectual property in a lawsuit can take significant time and money. We may not have the resources to enforce our intellectual property if a third party infringes an issued patent claim. Infringement lawsuits may require significant time and money resources. If we do not have such resources, for patents that we have licensed from a third party, the licensor is not obligated to help us enforce our patent rights. If the licensor does take action by filing a lawsuit claiming infringement, we will not be able to participate in the suit and therefore will not have control over the proceedings or the outcome of the suit. |
| ● | Because of the time, money and effort involved in obtaining and enforcing patents, our management may spend less time and resources on developing potential drug candidates than they otherwise would, which could increase our operating expenses and delay product programs. |
| ● | There can be no assurance that any of our patent applications, including any licensed patent applications, will result in the issuance of patents, and we cannot predict the breadth of claims that may be allowed in our currently pending patent applications or in patent applications we may file or license from others in the future. |
| ● | Issuance of a patent may not provide much practical protection. If we receive a patent of narrow scope, then it may be easy for competitors to design products that do not infringe our patent(s). |
| ● | We have limited ability to expand coverage of our licensed patent related to SHLP-2 and our licensed patent application related to SHLP-6 outside of the United States. The lack of patent protection in international jurisdictions may inhibit our ability to advance our drug candidates in these markets. |
| ● | If a court decides that the method of manufacture or use of any of our drug candidates infringes on a third-party patent, we may have to pay substantial damages for infringement. |
31
| ● | A court may prohibit us from making, selling or licensing a potential drug candidate unless the patent holder grants a license. A patent holder is not required to grant a license. If a license is available, we may have to pay substantial royalties or grant cross licenses to our patents, and the license terms may be unacceptable. |
| ● | Redesigning our potential drug candidates so that they do not infringe on other patents may not be possible or could require substantial funds and time. |
It is also unclear whether our trade secrets are adequately protected. While we use reasonable efforts to protect our trade secrets, our employees or consultants may unintentionally or willfully disclose our information to competitors. Enforcing a claim that someone illegally obtained and is using our trade secrets is expensive and time consuming, and the outcome is unpredictable. In addition, courts outside the United States are sometimes less willing to protect trade secrets. Our competitors may independently develop equivalent knowledge, methods and know-how. We may also support and collaborate in research conducted by government organizations, hospitals, universities or other educational institutions. These research partners may be unable or unwilling to grant us exclusive rights to technology or products derived from these collaborations prior to entering into the relationship.
If we do not obtain required intellectual property rights, we could encounter delays in our drug development efforts while we attempt to design around other patents or even be prohibited from developing, manufacturing or selling potential drug candidates requiring these rights or licenses. There is also a risk that disputes may arise as to the rights to technology or potential drug candidates developed in collaboration with other parties.
General Risk Factors
If we fail to establish and maintain proper and effective internal control over financial reporting in the future, our ability to produce accurate and timely financial statements could be impaired, which could harm our operating results, investors’ views of us and, as a result, the value of our common stock.
The Sarbanes-Oxley Act requires, among other things, that we maintain effective internal controls for financial reporting and disclosure controls and procedures and that we furnish a report by management on, among other things, the effectiveness of our internal control over financial reporting. This assessment needs to include disclosure of any material weaknesses identified by our management in our internal control over financial reporting. A material weakness is a deficiency, or combination of deficiencies, in internal control over financial reporting that results in more than a reasonable possibility that a material misstatement of annual or interim financial statements will not be prevented or detected on a timely basis. Section 404 of the Sarbanes-Oxley Act also generally requires an attestation from our independent registered public accounting firm on the effectiveness of our internal control over financial reporting. However, for as long as we are not an accelerated filer or large accelerated filer, we intend to take advantage of the exemption permitting us not to comply with the independent registered public accounting firm attestation requirement.
Our compliance with Section 404 will require us to document and evaluate our internal control over financial reporting, which is both costly and challenging. In this regard, we will need to continue to dedicate internal resources, potentially engage outside consultants and adopt a detailed work plan to assess and document the adequacy of internal control over financial reporting, continue steps to improve control processes as appropriate, validate through testing that controls are functioning as documented and implement a continuous reporting and improvement process for internal control over financial reporting. Despite our efforts, there is a risk that we will not be able to conclude that our internal control over financial reporting is effective as required by Section 404. If we identify one or more material weaknesses, it could result in an adverse reaction in the financial markets due to a loss of confidence in the reliability of our consolidated financial statements. In addition, if we are not able to continue to meet these requirements, we may not be able to remain listed on The Nasdaq Capital Market (“Nasdaq”).
As we continue to grow, we expect to hire additional personnel and may utilize external temporary resources to implement, document and modify policies and procedures to maintain effective internal controls. However, it is possible that we may identify deficiencies and weaknesses in our internal controls. If material weaknesses or deficiencies in our internal controls exist and go undetected or unremediated, our consolidated financial statements could contain material misstatements that, when discovered in the future, could cause us to fail to meet our future reporting obligations and cause the price of our common stock to decline.
32
Significant disruptions of information technology systems or security breaches could adversely affect our business.
We are increasingly dependent upon information technology systems, infrastructure and data to operate our business. In the ordinary course of business, we collect, store and transmit large amounts of confidential information (including, among other things, trade secrets or other intellectual property, proprietary business information and personal information). It is critical that we do so in a secure manner to maintain the confidentiality and integrity of such confidential information. We also have outsourced elements of our operations to third parties, and as a result we manage a number of third-party vendors who may or could have access to our confidential information. Attacks on information technology systems are increasing in their frequency, levels of persistence, sophistication and intensity, and they are being conducted by increasingly sophisticated and organized groups and individuals with a wide range of motives and expertise. The size and complexity of our information technology systems, and those of third-party vendors with whom we contract, and the large amounts of confidential information stored on those systems, make such systems vulnerable to service interruptions or to security breaches from inadvertent or intentional actions by our employees, third-party vendors, and/or business partners, or from cyber-attacks by malicious third parties. Cyber-attacks could include the deployment of harmful malware, ransomware, denial-of-service attacks, social engineering and other means to affect service reliability and threaten the confidentiality, integrity and availability of information.
Significant disruptions of our information technology systems, or those of our third-party vendors, or security breaches could adversely affect our business operations and/or result in the loss, misappropriation and/or unauthorized access, use or disclosure of, or the prevention of access to, confidential information, including, among other things, trade secrets or other intellectual property, proprietary business information and personal information, and could result in financial, legal, business and reputational harm to us.
Any failure or perceived failure by us or any third-party collaborators, service providers, contractors or consultants to comply with our privacy, confidentiality, data security or similar obligations to third parties, or any data security incidents or other security breaches that result in the unauthorized access, release or transfer of sensitive information, including personally identifiable information, may result in governmental investigations, enforcement actions, regulatory fines, litigation or public statements against us, could cause third parties to lose trust in us or could result in claims by third parties asserting that we have breached our privacy, confidentiality, data security or similar obligations, any of which could have a material adverse effect on our reputation, business, financial condition or results of operations. Moreover, data security incidents and other security breaches can be difficult to detect, and any delay in identifying them may lead to increased harm. While we have implemented data security measures intended to protect our information technology systems and infrastructure, there can be no assurance that such measures will successfully prevent service interruptions or data security incidents.
If securities or industry analysts do not publish or cease publishing research or reports about us, our business or our market, or if they change their recommendations regarding our stock adversely, our stock price and trading volume could decline.
The trading market for our common stock will be influenced by the research and reports that industry or securities analysts may publish about us, our business, our market or our competitors. If any of the analysts who may cover us change their recommendation regarding our stock adversely, or provide more favorable relative recommendations about our competitors, our stock price would likely decline. If any analysts who may cover us were to cease coverage of our Company or fail to regularly publish reports on us, we could lose visibility in the financial markets, which in turn could cause our stock price or trading volume to decline.
The price of our common stock may be volatile and fluctuate substantially, which could result in substantial losses for holders of our common stock.
The market price of our common stock has been and is likely to continue to be volatile. The stock market in general, and the market for biotechnology companies in particular has experienced extreme volatility that has often been unrelated to the operating performance of particular companies. The market price for our common stock may be influenced by many factors, including:
| ● | results of preclinical studies or clinical trials of our product candidates or those of our competitors: |
| ● | unanticipated or serious safety concerns related to the use of any of our product candidates; |
| ● | adverse regulatory decisions, including failure to receive regulatory approval for any of our product candidates; |
33
| ● | the success of competitive drugs or technologies; |
| ● | regulatory or legal developments in the United States and other countries applicable to our product candidates; |
| ● | the size and growth of our prospective patient populations; |
| ● | developments concerning our collaborators, our external manufacturers or in-house manufacturing capabilities; |
| ● | inability to obtain adequate product supply for any product candidate for preclinical studies, clinical trials or future commercial sale or inability to do so at acceptable prices; |
| ● | developments or disputes concerning patent applications, issued patents or other proprietary rights; |
| ● | the recruitment or departure of key personnel; |
| ● | the level of expenses related to any of our product candidates or clinical development programs; |
| ● | the results of our efforts to discover, develop, acquire or in-license additional product candidates or drugs; |
| ● | actual or anticipated changes in estimates as to financial results, development timelines or recommendations by securities analysts or publications of research reports about us or our industry; |
| ● | variations in our financial results or those of companies that are perceived to be similar to us; |
| ● | changes in the structure of healthcare payment systems; |
| ● | market conditions in the biotechnology sector; |
| ● | our cash position or the announcement or expectation of additional financing efforts; |
| ● | general economic, industry and market conditions; and |
| ● | other factors, including those described in this “Risk Factors” section, many of which are beyond our control. |
The price of our common stock does not meet the requirements for continued listing on Nasdaq. If we fail to regain compliance with the minimum listing requirements, our common stock will be subject to delisting. Our ability to publicly or privately sell equity securities and the liquidity of our common stock could be adversely affected if our common stock is delisted.
The continued listing standards of Nasdaq require, among other things, that the minimum bid price of a listed company’s stock be at or above $1.00. If the closing minimum bid price is below $1.00 for a period of more than 30 consecutive trading days, the listed company will fail to be in compliance with Nasdaq’s listing rules and, if it does not regain compliance within the grace period, will be subject to delisting. As previously reported, on November 10, 2021, we received a notice from the Nasdaq Listing Qualifications Department notifying us that for 30 consecutive trading days, the bid price of our common stock had closed below the minimum $1.00 per share requirement. In accordance with Nasdaq’s listing rules, we were afforded a grace period of 180 calendar days, or until May 9, 2022, to regain compliance with the bid price requirement. In order to regain compliance, the bid price of our common stock must close at a price of at least $1.00 per share for a minimum of 10 consecutive trading days.
If we fail to regain compliance by May 9, 2022, we may be eligible for a second 180 day compliance period, provided that, on such date, we meet the continued listing requirement for market value of publicly held shares and all other applicable Nasdaq listing requirements (other than the minimum closing bid price requirement) and we provide written notice to Nasdaq of our intention to cure the deficiency during the second compliance period, by effecting a reverse stock split, if necessary. Such extension of the grace period would be subject to Nasdaq’s discretion, and there can be no guarantee that we would be granted an extension.
34
We cannot provide any guarantee that we will regain compliance during the grace period or be able to maintain compliance with Nasdaq’s listing requirements in the future. If we are not able to regain compliance during the grace period, or any extension of the grace period for which we may be eligible, our common stock will be subject to delisting. Delisting from Nasdaq could adversely affect our ability to raise additional financing through the public or private sale of equity securities, would significantly affect the ability of investors to trade our securities and would negatively affect the value and liquidity of our common stock. Delisting could also have other negative results, including the potential loss of confidence by employees, the loss of institutional investor interest and fewer business development opportunities.
The requirements of being a public company may strain our resources, divert management’s attention and require us to disclose information that is helpful to competitors, make us more attractive to potential litigants and make it more difficult to attract and retain qualified personnel.
As a public company, we are subject to the reporting requirements of the Securities Act of 1933, as amended, the Exchange Act, the Sarbanes-Oxley Act, the Dodd-Frank Wall Street Reform and Consumer Protection Act of 2010, and applicable Canadian securities rules and regulations. Despite recent reforms made possible by the JOBS Act, compliance with these rules and regulations creates significant legal and financial compliance costs and makes some activities difficult, time-consuming or costly. The Exchange Act and applicable Canadian provincial securities legislation require, among other things, that we file annual, quarterly and current reports with respect to our business and operating results.
Additionally, the Sarbanes-Oxley Act and the related rules and regulations of the SEC and Nasdaq require us to implement particular corporate governance practices and adhere to a variety of reporting requirements and complex accounting rules. Among other things, we are subject to rules regarding the independence of the members of our board of directors and committees of the board and their experience in finance and accounting matters, rules regarding the diversity of our board of directors and certain of our executive officers are required to provide certifications in connection with our quarterly and annual reports filed with the SEC. The perceived personal risk associated with these rules may deter qualified individuals from accepting these positions. Accordingly, we may be unable to attract and retain qualified officers and directors. If we are unable to attract and retain qualified officers and directors, our business and our ability to maintain the listing of our shares of common stock on Nasdaq or another stock exchange could be adversely affected.
We are also subject to more stringent state law requirements. For example, under California law, we will be required to have at least three female directors on our board of directors and one director from an “underrepresented community” starting December 31, 2021, and two additional directors from an “underrepresented community” starting December 31, 2022. A director from an “underrepresented community” means a director who self-identifies as Black, African American, Hispanic, Latino, Asian, Pacific Islander, Native American, Native Hawaiian, Alaska Native, gay, lesbian, bisexual or transgender. If we fail to comply with either of these requirements, we could be fined by the California Secretary of State, our reputation may be adversely affected and certain investors may divest their holdings in our common stock.
Changes in U.S. federal income and other tax laws could adversely affect us.
New U.S. legislation or regulations which could affect our tax burden could be enacted by the U.S. government. We cannot predict the timing or extent of such tax-related developments which could have a negative impact on our financial results. Additionally, we use our best judgment in attempting to quantify and reserve for these tax obligations. However, a challenge by a taxing authority, our ability to utilize tax benefits such as carryforwards or tax credits, or a deviation from other tax-related assumptions could have a material adverse effect on our business, results of operations, or financial condition.
Unfavorable global economic conditions could adversely affect our business, financial condition or results of operations.
Our results of operations could be adversely affected by general conditions in the global economy and in the global financial markets. For example, the global financial crisis caused extreme volatility and disruptions in the capital and credit markets. A severe or prolonged economic downturn, such as a global financial crisis, could result in a variety of risks to our business, including, weakened demand for our product candidates and our ability to raise additional capital when needed on acceptable terms, if at all. A weak or declining economy could also strain our suppliers, possibly resulting in supply disruptions. Any of the foregoing could harm our business, and we cannot anticipate all of the ways in which the current economic climate and financial market conditions could adversely impact our business.
35
We or the third parties upon whom we depend may be adversely affected by natural disasters, and our business continuity and disaster recovery plans may not adequately protect us from a serious disaster.
Natural disasters could severely disrupt our operations and have a material adverse effect on our business, results of operations, financial condition and prospects. For example, our corporate headquarters are located in the San Francisco Bay Area, which has experienced both severe earthquakes and the effects of wildfires. We do not carry earthquake insurance. In addition, the long-term effects of climate change on general economic conditions and the biopharmaceutical industry in particular are unclear, and may heighten or intensify existing risk of natural disasters. If an earthquake, wildfire, other natural disaster, power outage or other event occurred that prevented us from using all or a significant portion of our headquarters, that damaged critical infrastructure or that otherwise disrupted operations, it may be difficult or, in certain cases, impossible for us to continue our business for a substantial period of time. The disaster recovery and business continuity plans we have in place may prove inadequate in the event of a serious disaster or similar event. We may incur substantial expenses as a result of the limited nature of our disaster recovery and business continuity plans, which could have a material adverse effect on our business.
Our employees, principal investigators, CROs and consultants may engage in misconduct or other improper activities, including non-compliance with regulatory standards and requirements and insider trading.
We are exposed to the risk of fraud or other misconduct by our employees, principal investigators, consultants and commercial partners. Misconduct by these parties could include intentional failures to comply with the regulations of FDA and non-U.S. regulators, provide accurate information to the FDA and non-U.S. regulators, comply with healthcare fraud and abuse laws and regulations in the United States and abroad, report financial information or data accurately or disclose unauthorized activities to us. In particular, sales, marketing and business arrangements in the healthcare industry are subject to extensive laws and regulations intended to prevent fraud, misconduct, kickbacks, self-dealing and other abusive practices. These laws and regulations may restrict or prohibit a wide range of pricing, discounting, marketing and promotion, sales commission, customer incentive programs and other business arrangements. Such misconduct could also involve the improper use of information obtained in the course of clinical studies, which could result in regulatory sanctions and cause serious harm to our reputation. We have adopted a code of ethics, but it is not always possible to identify and deter employee misconduct, and the precautions we take to detect and prevent this activity may not be effective in controlling unknown or unmanaged risks or losses or in protecting us from governmental investigations or other actions or lawsuits stemming from a failure to comply with these laws or regulations. If any such actions are instituted against us, and we are not successful in defending ourselves or asserting our rights, those actions could have a significant impact on our business, including the imposition of significant fines or other sanctions.
Item 1B. Unresolved Staff Comments
None.
Item 2. Properties
We are a party to a lease agreement for laboratory space leased on a month-to month basis that is part of a shared facility in Menlo Park, California. In September 2021, we renewed our lease for office space in Fairfield, New Jersey for an additional year at the same annual cost of $13,080 per annum.
Rent expense amounted to $0.4 million in each of the years ended December 31, 2021 and 2020.
Item 3. Legal Proceedings
From time to time, we may be subject to legal proceedings and claims in the ordinary course of business. We are not currently a party to any material legal proceedings, and to our knowledge none is threatened. There can be no assurance that future legal proceedings arising in the ordinary course of business or otherwise will not have a material adverse effect on our financial position, results of operations or cash flows.
Item 4. Mine Safety Disclosures
Not applicable.
36
PART II
Item 5. Market for Registrant’s Common Equity, Related Stockholder Matters and Issuer Purchases of Equity Securities
Market for our Common Stock
Our common stock has been trading on The Nasdaq Capital Market under the symbol “CWBR” since December 15, 2017.
Holders of Common Stock
As of March 24, 2022, there were 87 million shares of our common stock outstanding held by approximately 36 holders of record. A substantially greater number of holders of our common stock are “street name” or beneficial holders, whose shares of record are held by banks, brokers, and other financial institutions.
Dividends
We have not declared or paid a cash dividend on our capital stock and do not intend to pay cash dividends for the foreseeable future. All dividends are subject to the approval of our board of directors. Any future determinations to pay dividends on our capital stock would depend on our results of operations, our financial condition and liquidity requirements, restrictions that may be imposed by applicable laws or our contracts, and any other factors that our board of directors in its sole discretion may consider relevant in declaring a dividend.
Purchases of Equity Securities by the Issuer and Affiliated Purchasers
None.
Securities Authorized for Issuance under Equity Compensation Plans
See Item 12 for Equity Compensation Plan Information.
Recent Sales of Unregistered Securities
None.
Item 6. [Reserved]
37
Item 7. Management’s Discussion and Analysis of Financial Condition and Results of Operations
We are a clinical stage biotechnology company exploiting the power of the mitochondria and the peptides encoded in its genome to develop potential breakthrough therapeutics targeting chronic and age-related diseases. Our novel approach is built on the insight of our founders that certain mitochondrially encoded peptides produce effects that are not limited to local regulation within the mitochondria and may have important roles to play in critical systemic biological pathways quite distinct from what has traditionally been thought of as mitochondrial function.
We believe we have achieved a leading position in exploring the mitochondrial genome and its utility for the development of novel therapeutics, including world-renowned expertise in mitochondrial biology, a broad intellectual property estate with more than 65 patent applications filed, key opinion leaders and disciplined drug discovery and development processes. Our proprietary processes of identifying nucleic acid sequences encoding native peptides in the mitochondrial genome, developing and optimizing novel analogs of these natural mitochondrial derived peptides (“MDPs”), as well as developing and conducting proprietary screens to identify and characterize the activities of these peptides are referred to as our Mito+ platform. We are using our Mito+ platform to identify and develop novel modified versions of natural peptides, which we call analogs, to treat a variety of serious conditions, with a focus on diseases involving inflammation and fibrosis. We believe that the mitochondrial genome may be transformative in the field of drug discovery and that our novel peptide analogs may become a new and major class of drugs with broad therapeutic application.
We are currently advancing a pipeline of novel peptide analogs through varying stages of development. In August 2021, we announced positive topline data from a Phase 1a/1b clinical trial of CB4211, our most advanced compound, which is under development for the treatment of nonalcoholic steatohepatitis (“NASH”) and obesity. The study met its primary endpoint as CB4211 was well-tolerated and appeared safe with no serious adverse events. The evaluation of the exploratory endpoints in the Phase 1b portion of the trial, which was conducted in obese subjects with nonalcoholic fatty liver disease, showed significant reductions from baseline in key biomarkers of liver damage, ALT and AST, and in glucose levels in the CB4211 group compared to placebo after four weeks of treatment, with a trend towards lower body weight. We believe these positive clinical data are an important validation of our overall approach to drug discovery, serving as a proof point that novel analogs of peptides encoded in the mitochondrial genome can impact systemic biological pathways in humans while having an attractive safety and tolerability profile. Our second clinical candidate, CB5138-3, is a peptide with broad anti-fibrotic and anti-inflammatory properties. This program is currently in IND-enabling studies and we intend to pursue an initial indication of idiopathic pulmonary fibrosis (“IPF”). In addition, we have multiple preclinical programs, such as a program in acute respiratory distress syndrome (“ARDS”) as well as earlier stage discovery programs.
We have financed our operations primarily with proceeds from sales of our equity securities, including our initial public offering, private placements of our securities, a debt offering, public sales of our securities and the exercise of outstanding warrants and stock options. Since our inception through December 31, 2021, our operations have been funded with an aggregate of approximately $97.4 million from the sale and issuance of equity instruments and debt, including the proceeds from the exercise of warrants and stock options.
Since inception, we have incurred significant operating losses. Our net losses were $15.5 million and $16.3 million for the years ended December 31, 2021 and 2020, respectively. We incurred $2.7 million and $5.2 million in non-cash expenses during the years ended December 31, 2021 and 2020, respectively. Our net losses excluding non-cash expenses were $12.8 million and $11.1 million for the years ended December 31, 2021 and 2020, respectively. As of December 31, 2021, we had an accumulated deficit of $84.7 million. We expect to continue to incur significant expenses and operating losses over the next several years. Our net losses may fluctuate significantly from quarter to quarter and from year to year. Although we anticipate our research and development expenses to increase as we incur the costs related to our IND-enabling studies and potential initial clinical costs for our CB5138-3 program in addition to general program costs and the discovery and evaluation of other MDPs and optimization of novel analogs as potential drug candidates, the extent of that increase is uncertain at this time and subject to change due to the ongoing COVID-19 pandemic and other factors.
Impacts of the COVID-19 Pandemic
The extent of the impact of COVID-19 on our operational and financial performance will depend on certain developments, including the duration of the outbreak, impact on our preclinical and clinical studies including patient enrollment and retention, employee or industry events, and effect on our suppliers, service providers and manufacturers, all of which are uncertain and cannot be predicted. The COVID-19 pandemic and its adverse effects are prevalent in the locations where we, our contract research organizations (“CROs”), suppliers or third-party business partners conduct business and, as a result, we may experience more pronounced disruptions in our operations, liquidity, supply chain, facilities, and clinical trials. With respect to our clinical trials, we have experienced delays due to our clinical sites closing down during the pandemic, we have experienced delays in enrollment due to those closures and weather-related events in the state where our clinical sites are located. We may in the future experience more significant delays in enrollment, participant dosing, distribution of clinical trial materials, study monitoring and data analysis that could materially adversely impact our business, results of operations and overall financial performance in future periods. Specifically, we may experience impact from restrictions on travel and in-person meetings, delays in site activations and enrollment of clinical trials, prioritization of hospital resources toward pandemic effort, delays in review by the FDA and comparable foreign regulatory agencies, and disruptions in our supply chain for our product candidates. As of the filing date of this Form 10-K, the extent to which the COVID-19 pandemic may impact our financial condition, results of operations or guidance is uncertain. The effect of the COVID-19 pandemic will not be fully reflected in our results of operations and overall financial performance until future periods. See the section titled “Risk Factors” for further discussion of the possible impact of the COVID-19 pandemic on our business.
38
Financial Operations Review
Revenue
To date, we have not generated any revenue from product sales and do not expect to generate any revenue from the sale of products in the near future. In the future, we will seek to generate revenue from product sales, either directly or under any future licensing, development or similar relationship with a strategic partner.
Research and Development Expenses
Research and development expenses consist primarily of costs incurred for our research activities, including our drug discovery efforts, and the development of our product candidates, which include:
| ● | employee-related expenses including salaries, benefits and stock-based compensation expense; |
| ● | expenses incurred under agreements with third parties, including CROs that conduct research and development and preclinical activities on our behalf and the cost of consultants; |
| ● | the cost of laboratory equipment, supplies and manufacturing test materials; and |
| ● | depreciation and other personnel-related costs associated with research and product development. |
We record all research and development expenses as incurred.
Our Research Programs
Our research and development programs include activities in support of the clinical development of our most advanced program, CB4211, as well as the operation of our platform technology related to the discovery and development of novel therapeutics, evaluation of newly discovered natural sequences, design of novel improved analogs, evaluation of their therapeutic potential and optimization of their characteristics as potential drug development candidates. Depending on factors of capability, cost, efficiency and intellectual property rights, we conduct our research programs at our laboratory facility, or externally, pursuant to contractual arrangements with CROs or under collaborative arrangements with academic institutions.
The success of our research programs and the timing of those programs and the possible development of research peptides into drug candidates is highly uncertain. As such, at this time, we cannot reasonably estimate or know the nature, timing or estimated costs of the efforts that will be necessary to complete research and development of a commercial drug. We are also unable to predict when, if ever, we will receive material net cash inflows from our operations. This is due to the numerous risks and uncertainties associated with developing medicines, including the uncertainty of:
| ● | developing appropriate manufacturing processes and formulations; |
| ● | establishing an appropriate safety profile with toxicology studies; |
| ● | obtaining appropriate regulatory approval for conducting clinical trials; |
| ● | successfully designing, enrolling and completing clinical trials; |
| ● | receiving marketing approvals from applicable regulatory authorities; |
| ● | establishing commercial manufacturing capabilities or making arrangements with third-party manufacturers; |
39
| ● | obtaining and enforcing patent and trade secret protection for our product candidates; |
| ● | launching commercial sales of the products, if and when approved, whether alone or in collaboration with others; and |
| ● | maintaining an acceptable safety profile of the products following approval. |
A change in the outcome of any of these variables with respect to the development of any of our product candidates would significantly change the costs and timing associated with the development of that product candidate.
Research and development activities are central to our business model. Most of our potential drug candidates are in early stages of investigational research. Candidates in later stages of clinical development generally have higher development costs than those in earlier stages of clinical development, primarily due to the increased size and duration of later-stage clinical trials. We expect research and development costs to increase for the foreseeable future as we incur the costs related to our IND-enabling studies and potential initial clinical costs for our CB5138-3 program in addition to general program costs and the discovery and evaluation of other MDPs and optimization of novel analogs as potential drug candidates. However, we do not believe that it is possible at this time to accurately project total program-specific expenses through commercialization. There are numerous factors associated with the successful commercialization of any of our product candidates, including future trial design and various regulatory requirements, many of which cannot be determined with accuracy at this time based on our stage of development. Additionally, future commercial and regulatory factors beyond our control may impact our clinical development programs and plans.
General and Administrative Expenses
General and administrative expenses consist primarily of salaries and other related costs, including stock-based compensation, for personnel in executive, finance and administrative functions. Other significant costs include legal fees relating to patent and corporate matters and fees for accounting and consulting services and directors’ and officers’ insurance. We anticipate that our general and administrative expenses will remain relatively constant in the year ending December 31, 2022.
Results of Operations
The following tables set forth our results of operations for the periods presented. The year-to-year comparison of financial results is not necessarily indicative of financial results to be achieved in future periods.
| For The Years Ended December 31, | Change | |||||||||||||||
| 2021 | 2020 | $ | % | |||||||||||||
| Operating expenses: | ||||||||||||||||
| Research and development | $ | 7,705,090 | $ | 6,937,610 | $ | 767,480 | 11 | % | ||||||||
| General and administrative | 7,703,065 | 6,261,905 | 1,441,160 | 23 | % | |||||||||||
| Total operating expenses | $ | 15,408,155 | $ | 13,199,515 | $ | 2,208,640 | 17 | % | ||||||||
Comparison of Fiscal Years Ended December 31, 2021 and 2020
Operating Expenses
Research and development expenses were $7.7 million in the year ended December 31, 2021 compared to $6.9 million in the prior year, a $0.8 million increase, or 11%. The increase in research and development expenses in the year ended December 31, 2021, was primarily due to an increase of approximately $1.5 million associated with our research programs focused on continuing the development of our peptides primarily related to the costs of our current IND-enabling studies and the manufacture of cGMP drug substance. This increase was partially offset by a decrease of $0.5 million in clinical trial related costs due to the timing of those expenses and a $0.4 million decrease in stock-based compensation.
General and administrative expenses were $7.7 million in the year ended December 31, 2021 compared to $6.3 million in the prior year, a $1.4 million increase, or 23%. The increase in general and administrative expenses was due to higher compensation costs of approximately $1.0 million primarily related to higher stock-based compensation costs of $0.7 million and one-time charges related to the departure of our former CEO and a $0.2 million increase in insurance costs related to higher D&O premiums.
40
Liquidity and Capital Resources
As of December 31, 2021, we had $26.2 million in cash, cash equivalents and investments. As of December 31, 2020, we had $21.0 million in cash and cash equivalents. We maintain our cash in a checking and a savings account on deposit with a banking institution in the United States. Our cash equivalent balance as of December 31, 2021 and 2020 included $0.7 million and $0 million, respectively, of U.S. Treasury Bills that had maturity dates of less than three months at the date of purchase. As of December 31, 2021, we had working capital and stockholders’ equity of $25.3 million and $25.6 million, respectively, and incurred a net loss of $15.5 million for the year ended December 31, 2021.
In November 2021, we completed an underwritten public offering of our securities (the “2021 Public Offering”) pursuant to which we sold 20.8 million shares of our common stock and warrants to purchase 20.8 million shares of common stock for proceeds of $13.8 million, net of commissions and professional fees of approximately $1.2 million. The warrants issued in the 2021 Public Offering were immediately exercisable and have a term of five years and a per share exercise price of $0.72.
On May 27, 2020, we entered into an At-the-Market Offering Sales Agreement (“ATM”) with Virtu Americas, LLC, as sales agent, pursuant to which we may sell shares of common stock with an aggregate offering price of up to $20 million. During the year ended December 31, 2021, we had sold 1.7 million shares of our common stock under the ATM program for proceeds of $2.9 million, net of commissions and incurred professional fees of approximately $21,000. During the year ended December 31, 2020, we had sold 2.4 million shares of our common stock under the ATM program for proceeds of $4.3 million, net of commissions and professional fees of $0.2 million. As of December 31, 2021, we had $12.5 million available in our ATM program.
In August 2020, we completed an underwritten public offering of our securities (the “2020 Public Offering”) pursuant to which we sold 12.3 million shares of our common stock and warrants to purchase 10.6 million shares of common stock for proceeds of $13.7 million, net of commissions and professional fees of $1.4 million. The warrants issued in the 2020 Public Offering were immediately exercisable and have a term of five years and a per share exercise price of $1.44.
As reflected in the financial statements, we had an accumulated deficit as of December 31, 2021 and 2020, as well as recurring losses and negative cash flows from operating activities from inception. These factors raise substantial doubt about our ability to continue as a going concern for at least one year from the issuance of these financial statements. However, based on current budget assumptions, projected cash burn and our latitude to manage that cash burn and the cash and investments on hand as of December 31, 2021, we believe that we have sufficient capital to meet our operating expenses and obligations for the next twelve months from the date of this filing. However, if unanticipated difficulties or circumstances arise, we may require additional capital sooner to support our operations. If we are unable to raise additional capital whenever necessary, we may be forced to decelerate or curtail our research and development activities and/or other operations until such time as additional capital becomes available. Such limitation of our activities would allow us to slow our rate of spending and extend our use of cash until additional capital is raised. There can be no assurance that such a plan would be successful. There is no assurance that additional financing will be available when needed or that we will be able to obtain such financing on reasonable terms.
Cash Flows from Operating Activities
Net cash used in operating activities for the years ended December 31, 2021 and 2020 was $14.4 million and $9.8 million, respectively. Cash used in operations for the year ended December 31, 2021 was primarily due to our net loss of $15.5 million, which was partially offset by the non-cash item of stock based-compensation and decrease in accrued liabilities due to the timing of those expenses. Cash used in operations for the year ended December 31, 2020 was primarily due to our net loss of $16.3 million, which was partially offset by non-cash items of stock-based compensation, depreciation and amortization of the debt discount totaling $5.2 million.
Cash Flows from Investing Activities
Net cash used in investing activities for the year ended December 31, 2021 was $3.1 million and net cash used in investing activities for the year ended December 31, 2020 was $18.2 million. The cash used in investing activities for the years ended December 31, 2021 and 2020 was due to the timing of the purchases and maturities of our investments.
41
Cash Flows from Financing Activities
Net cash provided by financing activities for the years ended December 31, 2021 and 2020 was $19.7 million and $18.3 million, respectively. Cash provided by financing activities in the year ended December 31, 2021 was primarily due to net proceeds of $13.8 million from our underwritten public, $2.9 million received from our ATM offering, $2.1 million received from the exercise of warrants and $1.2 million from the exercise of employee stock options. Cash provided by financing activities in the year ended December 31, 2020 was due to net proceeds of $13.7 million and $4.3 million received from our underwritten public and ATM offering, respectively, and the exercise of stock options and warrants for total proceeds of $0.3 million.
Operating Leases
We are a party to a lease agreement for laboratory space leased on a month-to month basis that is part of a shared facility in Menlo Park, California. In September 2021, we renewed our lease for office space in Fairfield, New Jersey for an additional year at the same annual cost of $13,080 per annum.
Rent expense amounted to $0.4 million in each of the years ended December 31, 2021 and 2020.
Recent Accounting Pronouncements
See Note 3 “Summary of Significant Account Policies – Recent Accounting Pronouncements” to our Financial Statements for the year ended December 31, 2021, for a summary of the relevant recent accounting pronouncements.
Other recent accounting standards that have been issued or proposed by the FASB or other standards-setting bodies that do not require adoption until a future date are not expected to have a material impact on the Company’s financial statements upon adoption.
Critical Accounting Estimates
Our management’s discussion and analysis of our financial condition and results of operations are based on our financial statements, which have been prepared in accordance with accounting principles generally accepted in the United States (“U.S. GAAP”). U.S. GAAP requires us to make certain estimates and judgments that can affect the reported amounts of assets and liabilities as of the dates of the financial statements, the disclosure of contingencies as of the dates of the financial statements, and the reported amounts of revenue and expenses during the periods presented. We base our estimates on historical experience and on various other assumptions that we believe to be reasonable under the circumstances, the results of which form the basis for making judgments about the carrying values of assets and liabilities. If actual results or events differ materially from those contemplated by us in making these estimates, our reported financial condition and results of operations for future periods could be materially affected. See “Risk Factors” for certain matters that may affect our future financial condition or results of operations. An accounting policy is deemed to be critical if it requires an accounting estimate to be made based on assumptions about matters that are uncertain at the time the estimate is made, if different estimates reasonably could have been used, or if the changes in estimate that are reasonably likely to occur could materially impact the financial statements. Our management has discussed the development, selection and disclosure of these estimates with the audit committee of our board of directors.
The following critical accounting estimates reflect significant judgments and estimates used in the preparation of our financial statements:
| ● | Fair value of financial instruments |
| ● | Share-based payments |
| ● | Valuation of deferred tax assets |
42
Fair Value of Financial Instruments
We measure the fair value of financial assets and liabilities based on the exchange price that would be received for an asset or paid to transfer a liability (an exit price) in the principal or most advantageous market for the asset or liability in an orderly transaction between market participants on the measurement date. We utilize three levels of inputs that may be used to measure fair value:
| ● | Level 1 – quoted prices in active markets for identical assets or liabilities. |
| ● | Level 2 – quoted prices for similar assets and liabilities in active markets or inputs that are observable. |
| ● | Level 3 – inputs that are unobservable (for example, cash flow modeling inputs based on assumptions). |
The carrying amounts of cash, accounts payable, accrued liabilities and debt approximate fair value due to the short-term nature of these instruments.
Share-based Payments
We account for share-based payments using the fair value method. For employees and directors, the fair value of the award is measured on the grant date. For non-employees, fair value is generally measured based on the fair value of the services provided or the fair value of the common stock on the measurement date, whichever is more readily determinable. We have historically granted stock options at exercise prices no less than the fair market value as determined by the board of directors, with input from management.
See Note 3 “Summary of Significant Account Policies – Share-Based Payment” to our Financial Statements for the years ended December 31, 2021 and 2020 regarding the specific assumptions used with respect to stock-based compensation for the periods presented.
Valuation of Deferred Tax Assets
We recognize deferred tax assets and liabilities for the expected future tax consequences of items that have been included or excluded in the financial statements or tax returns. Deferred tax assets and liabilities are determined on the basis of the difference between the tax basis of assets and liabilities and their respective financial reporting amounts (“temporary differences”) at enacted tax rates in effect for the years in which the temporary differences are expected to reverse.
The benefit of tax positions taken or expected to be taken in income tax returns are recognized in the financial statements if such positions are more likely than not of being sustained. We have evaluated and concluded that there were no material uncertain tax positions requiring recognition in the Company’s financial statements as of December 31, 2021 and 2020. The Company does not expect any significant changes in the unrecognized tax benefits within twelve months of the reporting date.
Item 7A. Quantitative and Qualitative Disclosures about Market Risk
Not applicable.
43
Item 8. Financial Statements
F-1
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
To the Shareholders and Board of Directors of
CohBar, Inc.
Opinion on the Financial Statements
We have audited the accompanying balance sheets of CohBar, Inc. (the “Company”) as of December 31, 2021 and 2020, the related statements of operations, changes in stockholders’ equity and cash flows for each of the two years in the period ended December 31, 2021, and the related notes (collectively referred to as the “financial statements”). In our opinion, the financial statements present fairly, in all material respects, the financial position of the Company as of December 31, 2021 and 2020, and the results of its operations and its cash flows for each of the two years in the period ended December 31, 2021, in conformity with accounting principles generally accepted in the United States of America.
Basis for Opinion
These financial statements are the responsibility of the Company’s management. Our responsibility is to express an opinion on the Company’s financial statements based on our audits. We are a public accounting firm registered with the Public Company Accounting Oversight Board (United States) (“PCAOB”) and are required to be independent with respect to the Company in accordance with the U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB.
We conducted our audits in accordance with the standards of the PCAOB. Those standards require that we plan and perform the audits to obtain reasonable assurance about whether the financial statements are free of material misstatement, whether due to error or fraud. The Company is not required to have, nor were we engaged to perform, an audit of its internal control over financial reporting. As part of our audits we are required to obtain an understanding of internal control over financial reporting but not for the purpose of expressing an opinion on the effectiveness of the Company’s internal control over financial reporting. Accordingly, we express no such opinion.
Our audits included performing procedures to assess the risks of material misstatement of the financial statements, whether due to error or fraud, and performing procedures that respond to those risks. Such procedures included examining, on a test basis, evidence regarding the amounts and disclosures in the financial statements. Our audits also included evaluating the accounting principles used and significant estimates made by management, as well as evaluating the overall presentation of the financial statements. We believe that our audits provide a reasonable basis for our opinion.
Critical Audit Matters
Critical Audit Matters are matters arising from the current period audit of the financial statements that were communicated or required to be communicated to the audit committee and that: (1) relate to accounts or disclosures that are material to the financial statements and (2) involved our especially challenging, subjective, or complex judgments. We determined that there are no critical audit matters.
/s/ Marcum llp
We have served as the Company’s auditor since 2014
March 29, 2022
F-2
CohBar, Inc.
Balance Sheets
| As of | ||||||||
| December 31, 2021 | December 31, 2020 | |||||||
| ASSETS | ||||||||
| Current assets: | ||||||||
| Cash and cash equivalents | $ | $ | ||||||
| Investments | ||||||||
| Vendor receivable | ||||||||
| Prepaid expenses and other current assets | ||||||||
| Total current assets | ||||||||
| Property and equipment, net | ||||||||
| Intangible assets, net | ||||||||
| Other assets | ||||||||
| Total assets | $ | $ | ||||||
| LIABILITIES AND STOCKHOLDERS’ EQUITY | ||||||||
| Current liabilities: | ||||||||
| Accounts payable | $ | $ | ||||||
| Accrued liabilities | ||||||||
| Accrued payroll and other compensation | ||||||||
| Note payable, net of debt discount and offering costs of $ | ||||||||
| Total current liabilities | ||||||||
| Notes payable, net of debt discount and offering costs of $ | ||||||||
| Total liabilities | ||||||||
| Commitments and contingencies | ||||||||
| Stockholders’ equity: | ||||||||
| No shares issued and outstanding as of December 31, 2021 and December 31, 2020, respectively | - | - | ||||||
| Common stock, $0.001 par value, Authorized 180,000,000 shares; | ||||||||
| Issued and outstanding | ||||||||
| Additional paid-in capital | ||||||||
| Accumulated deficit | ( | ) | ( | ) | ||||
| Total stockholders’ equity | ||||||||
| Total liabilities and stockholders’ equity | $ | $ | ||||||
The accompanying notes are an integral part of these financial statements
F-3
CohBar, Inc.
Statement of Operations
For The Years Ended December 31, | ||||||||
| 2021 | 2020 | |||||||
| Revenues | $ | $ | ||||||
| Operating expenses: | ||||||||
| Research and development | ||||||||
| General and administrative | ||||||||
| Total operating expenses | ||||||||
| Operating loss | ( | ) | ( | ) | ||||
| Other income (expense): | ||||||||
| Interest income | ||||||||
| Interest expense | ( | ) | ( | ) | ||||
| Equity modification expense | ( | ) | ||||||
| Amortization of debt discount and offering costs | ( | ) | ( | ) | ||||
| Total other expense | ( | ) | ( | ) | ||||
| Net loss | $ | ( | ) | $ | ( | ) | ||
| Basic and diluted net loss per share | $ | ( | ) | $ | ( | ) | ||
| Weighted average common shares outstanding - basic and diluted | ||||||||
The accompanying notes are an integral part of these financial statements
F-4
CohBar, Inc.
Statements of Changes in Stockholders’ Equity
| Common Stock | Additional Paid-in- | Accumulated | Stockholders’ | |||||||||||||||||
| Number | Amount | Captial | Deficit | Equity | ||||||||||||||||
| Balance, December 31, 2019 | $ | $ | $ | ( | ) | |||||||||||||||
| Stock-based compensation | - | |||||||||||||||||||
| Equity modification expense | - | |||||||||||||||||||
| Exercise of employee stock options | ||||||||||||||||||||
| Exercise of warrants | ||||||||||||||||||||
| Sale of common stock in ATM, net | ||||||||||||||||||||
| Sale of common stock in CMPO, net | ||||||||||||||||||||
| Issuance of equity to convert debt | ||||||||||||||||||||
| Net loss | - | ( | ) | ( | ) | |||||||||||||||
| Balance, December 31, 2020 | $ | $ | $ | ( | ) | $ | ||||||||||||||
| Stock-based compensation | - | |||||||||||||||||||
| Issuance of common stock for ESPP plan | ||||||||||||||||||||
| Exercise of employee stock options | ||||||||||||||||||||
| Exercise of warrants | ||||||||||||||||||||
| Sale of common stock in ATM, net | ||||||||||||||||||||
| Sale of common stock in CMPO, net | ||||||||||||||||||||
| Net loss | - | ( | ) | ( | ) | |||||||||||||||
| Balance, December 31, 2021 | $ | $ | $ | ( | ) | |||||||||||||||
The accompanying notes are an integral part of these financial statements
F-5
CohBar, Inc.
Statements
of Cash Flows
| For The Years Ended December 31, | ||||||||
| 2021 | 2020 | |||||||
| Cash flows from operating activities: | ||||||||
| Net loss | $ | ( | ) | $ | ( | ) | ||
| Adjustments to reconcile net loss to net cash used in operating activities: | ||||||||
| Depreciation and amortization | ||||||||
| Stock-based compensation | ||||||||
| Equity modification expense | ||||||||
| Amortization of debt discount | ||||||||
| Amortization of debt issuance costs | ||||||||
| Discount on investments | ( | ) | ||||||
| Changes in operating assets and liabilities: | ||||||||
| Vendor receivable | ( | ) | ||||||
| Prepaid expenses and other current assets | ( | ) | ( | ) | ||||
| Accounts payable | ( | ) | ||||||
| Accrued liabilities | ( | ) | ||||||
| Accrued payroll and other compensation | ( | ) | ||||||
| Net cash used in operating activities | ( | ) | ( | ) | ||||
| Cash flows from investing activities: | ||||||||
| Purchases of property and equipment | ( | ) | ( | ) | ||||
| Payment for security deposit | ( | ) | ( | ) | ||||
| Patent costs | ( | ) | ||||||
| Purchases of investments | ( | ) | ( | ) | ||||
| Proceeds from redemptions of investments | ||||||||
| Net cash used in investing activities | ( | ) | ( | ) | ||||
| Cash flows from financing activities: | ||||||||
| Proceeds from ESPP plan | ||||||||
| Proceeds from public offering | ||||||||
| Costs of public offering | ( | ) | ( | ) | ||||
| Proceeds from the At-the-Market Offering | ||||||||
| Costs of At-the-Market Offering | ( | ) | ( | ) | ||||
| Proceeds from exercise of warrants | ||||||||
| Repayment of promissory notes | ( | ) | ||||||
| Proceeds from exercise of employee stock options | ||||||||
| Net cash provided by financing activities | ||||||||
| Net increase (decrease) in cash and cash equivalents | ( | ) | ||||||
| Cash and cash equivalents at beginning of period | ||||||||
| Cash and cash equivalents at end of period | $ | $ | ||||||
| Non-cash financing activities: | ||||||||
| Conversion of Promissory Notes to Common Stock | $ | $ | ||||||
| Supplemental disclosure of cash flow information: | ||||||||
| Cash paid for income taxes | $ | $ | ||||||
| Cash paid for interest | $ | $ | ||||||
The accompanying notes are an integral part of these financial statements
F-6
CohBar, Inc.
Notes to Financial Statements
Note 1 – Business Organization and Nature of Operations
CohBar, Inc. (“CohBar,” “its” or the “Company”) is a clinical stage biotechnology company exploiting the power of the mitochondria and the peptides encoded in its genome to develop potential breakthrough therapeutics targeting chronic and age-related diseases.
The Company’s primary activities include utilizing its MITO+ platform to identify and develop novel peptide analogs, the research and development of its pipeline, securing intellectual property protection for its discoveries and assets, managing collaborations and clinical trials with contract research organizations (“CROs”) and raising capital to fund the Company’s operations. To date, the Company has not generated any revenues from operations and does not expect to generate any revenues in the near future. The Company has financed its operations primarily with proceeds from sales of its equity securities, private placements, the exercise of outstanding warrants and stock options and the issuance of debt instruments.
The Company is monitoring the COVID-19 pandemic, which continues to rapidly evolve, and has taken steps to mitigate the potential impacts on its business. The extent to which the pandemic may impact the Company’s business, preclinical studies and its clinical trial will depend on future developments, which are highly uncertain and cannot be predicted with confidence. The Company has modified its business practices by restricting nonessential travel, implementing a partial work from home policy for its employees and instituting new safety protocols for its lab to enable essential on-site work to continue. The Company expects to continue to take actions that are in the best interests of its employees and business partners. Due to the uncertainty surrounding the pandemic, the Company’s visibility into the duration of these actions is limited.
Note 2 – Liquidity and Management’s Plans
As of December 31, 2021, the Company had a cash, cash equivalents and
investments balance of $
Note 3 – Summary of Significant Accounting Policies
Basis of Presentation
All amounts are presented in U.S. Dollars.
Use of Estimates
The preparation of financial statements in conformity with accounting principles generally accepted in the United States of America (“U.S. GAAP”) requires management to make estimates and assumptions that affect the reported amounts of assets and liabilities and disclosure of contingent liabilities at dates of the financial statements and the reported amounts of revenue and expenses during the periods. Actual results could differ from these estimates. The Company’s significant estimates and assumptions include the fair value of financial instruments, stock-based compensation and the valuation allowance relating to the Company’s deferred tax assets.
F-7
CohBar, Inc.
Notes to Financial Statements
Note 3 – Summary of Significant Accounting Policies (continued)
Concentrations of Credit Risk
The Company maintains deposits in a financial institution which is insured by the Federal Deposit Insurance Corporation (“FDIC”). At various times, the Company has deposits in this financial institution in excess of the amount insured by the FDIC. The Company has not experienced any losses in such accounts and believes it is not exposed to any significant credit risk.
Investments
Investments
as of December 2021 and 2020 consist of U.S. Treasury Bills, which are classified as held-to-maturity, and Certificates of Deposit
totaling $
Capitalization of Patent Costs
The
Company capitalizes the costs of its patents which consists of legal and filing fees related to the prosecution of patent filings.
The patents will be amortized using the straight-line method over the estimated remaining lives of the patents which is
Cash Equivalents
The
Company considers all highly liquid investments with an original maturity of three months or less when purchased to be cash
equivalents. As of December 31, 2021, the Company invested $
Property and Equipment, net
Property
and equipment are stated at cost less accumulated depreciation. Depreciation of computer and lab equipment is computed by use
of the straight-line method based on the estimated useful lives of the assets, which range from
Fair Value of Financial Instruments
The Company measures the fair value of financial assets and liabilities based on the exchange price that would be received for an asset or paid to transfer a liability (an exit price) in the principal or most advantageous market for the asset or liability in an orderly transaction between market participants on the measurement date. The Company utilizes three levels of inputs that may be used to measure fair value:
| Level 1 - | quoted prices in active markets for identical assets or liabilities. |
| Level 2 - | quoted prices for similar assets and liabilities in active markets or inputs that are observable. |
| Level 3 - | inputs that are unobservable (for example, cash flow modeling inputs based on assumptions). |
The carrying amounts of cash, investments and accounts payable approximate fair value due to the short-term nature of these instruments. The amount of debt included in the accompanying balance sheets approximates its fair value because the interest rate of the notes approximates the current market interest rate.
F-8
CohBar, Inc.
Notes to Financial Statements
Note 3 – Summary of Significant Accounting Policies (continued)
Common Stock Purchase Warrants
The Company classifies as equity any contracts that (i) require physical settlement or net-share settlement or (ii) provides the Company with a choice of net-cash settlement or settlement in its own shares (physical settlement or net-share settlement) providing that such contracts are indexed to the Company’s own stock. The Company classifies as assets or liabilities any contracts that (i) require net-cash settlement (including a requirement to net cash settle the contract if an event occurs and if that event is outside the Company’s control), or (ii) gives the counterparty a choice of net-cash settlement or settlement in shares (physical settlement or net-share settlement). The Company assesses classification of its common stock purchase warrants and other free-standing derivatives at each reporting date to determine whether a change in classification between assets, liabilities and equity is required. The Company’s free-standing derivatives consist of warrants to purchase common stock that were issued in connection with its notes payable and public and private offerings. The Company evaluated these warrants to assess their proper classification using the applicable criteria enumerated under U.S. GAAP and determined that the common stock purchase warrants meet the criteria for equity classification in the accompanying balance sheets as of December 31, 2021 and 2020.
Income Taxes
The Company recognizes deferred tax assets and liabilities for the expected future tax consequences of items that have been included or excluded in the financial statements or tax returns. Deferred tax assets and liabilities are determined on the basis of the difference between the tax basis of assets and liabilities and their respective financial reporting amounts (“temporary differences”) at enacted tax rates in effect for the years in which the temporary differences are expected to reverse.
The benefit of tax positions taken or expected to be taken in income tax returns are recognized in the financial statements if such positions are more likely than not of being sustained. Management has evaluated and concluded that there were no material uncertain tax positions requiring recognition in the Company’s financial statements as of December 31, 2021 and 2020. The Company does not expect any significant changes in the unrecognized tax benefits within twelve months of the reporting date.
The Company classifies interest expense and any related penalties related to income tax uncertainties as a component of income tax expense. No interest or penalties have been recognized during the years ended December 31, 2021 and 2020.
Research and Development Expenses
The Company expenses all research and development expenses as incurred. These costs include payroll, employee benefits, supplies, contracted for lab services, depreciation and other personnel-related costs associated with product development.
Share-Based Payment
The Company accounts for share-based payments using the fair value method. For employees and directors, the fair value of the award is measured, as discussed below, on the grant date. For non-employees, fair value is generally valued based on the fair value of the services provided or the fair value of the equity instruments on the measurement date, whichever is more readily determinable. The Company has granted stock options at exercise prices equal to the closing price of the Company’s common stock as reported by Nasdaq, with input from management on the date of grant. Upon exercise of an option or warrant, the Company issues new shares of common stock out of its authorized shares.
The weighted-average fair value of options and warrants has been estimated on the grant date or measurement date using the Black-Scholes pricing model. The fair value of each instrument is estimated on the grant date or measurement date utilizing certain assumptions for a risk-free interest rate, volatility and expected remaining lives of the awards. The risk-free interest rate used is the United States Treasury rate for the day of the grant having a term equal to the life of the equity instrument. Volatility was derived from the Company’s share price. The assumptions used in calculating the fair value of share-based payment awards represent management’s best estimates, but these estimates involve inherent uncertainties and the application of management judgment. As a result, if factors change and the Company uses different assumptions, the Company’s stock-based compensation expense could be materially different in the future.
F-9
CohBar, Inc.
Notes to Financial Statements
Note 3 – Summary of Significant Accounting Policies (continued)
The Black-Scholes assumptions are as follows:
| For the Years Ended December 31, | ||||||
| 2021 | 2020 | |||||
| Expected life | ||||||
| Risk free interest rate | ||||||
| Expected volatility | ||||||
| Expected dividend yield | ||||||
As
of December 31, 2021, total unrecognized stock compensation expense was $
Net Loss Per Share of Common Stock
Basic net loss per share is computed by dividing net loss attributable to common stockholders by the weighted average number of common shares outstanding during the period. Diluted net earnings per share reflects the potential dilution that could occur if securities or other instruments to issue common stock were exercised or converted into common stock. Potentially dilutive securities are excluded from the computation of diluted net loss per share as their inclusion would be anti-dilutive and consist of the following:
| As of December 31, | ||||||||
| 2021 | 2020 | |||||||
| Options | ||||||||
| Warrants | ||||||||
| Totals | ||||||||
Recent Accounting Pronouncements
In December 2019, the Financial Accounting Standards Board issued ASU No. 2019-12, “Income Taxes (Topic 740): Simplifying the Accounting for Income Taxes” (“ASU 2019-12”), which is intended to simplify the accounting for income taxes. ASU 2019-12 removes certain exceptions to the general principles in Topic 740. This guidance is effective for fiscal years, and interim periods within those fiscal years, beginning after December 15, 2020, with early adoption permitted. Upon adoption, ASU No. 2019-12 did not have an impact on the Company’s consolidated financial statements and related disclosures.
F-10
CohBar, Inc.
Notes to Financial Statements
Note 4 – Property and Equipment
Property and equipment consist of the following:
| As of December 31, | ||||||||
| 2021 | 2020 | |||||||
| Lab equipment | $ | $ | ||||||
| Computer and equipment | ||||||||
| Total property and equipment | $ | $ | ||||||
| Less: accumulated depreciation | ( | ) | ( | ) | ||||
| Total property and equipment, net | $ | $ | ||||||
Depreciation
expense related to property and equipment for the years ended December 31, 2021 and 2020 was $
Note 5 – Intangible Assets
Intangible assets consist of the following:
| As of December 31, | ||||||||
| 2021 | 2020 | |||||||
| Intangible assets: patents | $ | $ | ||||||
| Less: amortization | ( | ) | ( | ) | ||||
| Total intangible assets, net | $ | $ | ||||||
Amortization
expense for each of the years ended December 31, 2021 and 2020 was $
The
Company will recognize intangible amortization expense of $
Note 6 – Accrued Liabilities
Accrued liabilities consist of the following:
| As of December 31, | ||||||||
| 2021 | 2020 | |||||||
| Lab services & supplies | $ | $ | ||||||
| Professional fees | ||||||||
| Interest | ||||||||
| Other | ||||||||
| Total accrued liabilities | $ | $ | ||||||
F-11
CohBar, Inc.
Notes to Financial Statements
Note 7 – Notes Payable
During
the year ended December 31, 2020, the Company completed a private offering (the “Private Offering”) with certain
promissory note holders converting outstanding amounts due in 2021 and 2022 under its
Note 8 – Commitments and Contingencies
Litigations, Claims and Assessments
The Company may from time to time be a party to litigation and subject to claims incident to the ordinary course of business. As the Company grows and gains prominence in the marketplace it may become a party to an increasing number of litigation matters and claims. The outcome of litigation and claims cannot be predicted with certainty, and the resolution of these matters could materially affect the Company’s future results of operations, cash flows or financial position. The Company is not currently a party to any legal proceedings.
Licensing Agreements
The
Company is a party to an Exclusive License Agreement (the “2011 Exclusive Agreement”) with the Regents of the University
of California (“the Regents” or “Licensors”) which remains in effect for the life of the last-to-expire
patent or last to be abandoned patent application, whichever is later. The Company agreed to pay the Licensors specified development
milestone payments aggregating up to $
The
Company is also a party to an Exclusive License Agreement (the “2013 Exclusive Agreement”) with the Regents whereby
the Regents granted to the Company an exclusive license for the use of certain other patents. The 2013 Exclusive Agreement
remains in effect for the life of the last-to-expire patent or last to be abandoned patent application, whichever is later. The
Company paid the Regents an initial license issue fee of $
F-12
CohBar, Inc.
Notes to Financial Statements
Note 8 – Commitments and Contingencies (continued)
Operating Leases
The
Company is a party to a lease agreement for laboratory space leased on a month-to-month basis that is part of a shared facility
in Menlo Park, California. In September 2021, the Company renewed its lease for office space in Fairfield, New Jersey for an additional
year at the same annual cost of $
Rent
expense amounted to $
Note 9 – Income Taxes
The tax effects of temporary differences that give rise to deferred tax assets are as follows:
| As of December 31, | ||||||||
| 2021 | 2020 | |||||||
| Current: | ||||||||
| Accrued expenses | $ | $ | ||||||
| Stock compensation | ||||||||
| Net operating loss carryforward | ||||||||
| Research and development credit carry forward | ||||||||
| Total deferred tax assets | ||||||||
| Valuation allowance | ( | ) | ( | ) | ||||
| Deferred tax asset, net of valuation allowance | $ | $ | ||||||
A reconciliation of the statutory federal income tax rate to the Company’s effective tax rate is as follows:
| For the Years Ended December 31, | ||||||||
| 2021 | 2020 | |||||||
| U.S. statutory federal rate | ( | )% | ( | )% | ||||
| State income taxes, net of federal tax | ( | )% | ( | )% | ||||
| Federal tax rate change | % | % | ||||||
| Permanent differences | % | % | ||||||
| Prior year true-ups | % | ( | )% | |||||
| R&D tax credit | ( | )% | ( | )% | ||||
| Change in valuation allowance | % | % | ||||||
| Income tax provision (benefit) | % | % | ||||||
F-13
CohBar, Inc.
Notes to Financial Statements
Note 9 – Income Taxes (continued)
The income tax provision consists of the following:
| For the Years Ended December 31, | ||||||||
| 2021 | 2020 | |||||||
| Federal | ||||||||
| Current | $ | $ | ||||||
| Deferred | ( | ) | ( | ) | ||||
| State and local | ||||||||
| Current | ||||||||
| Deferred | ( | ) | ( | ) | ||||
| Change in valuation allowance | ||||||||
| Income tax provision (benefit) | $ | $ | ||||||
The
Company assesses the likelihood that deferred tax assets will be realized. To the extent that realization is not more-likely-than-not,
a valuation allowance is established. Based upon the Company’s losses since inception, management believes that it is more-likely-than-not
that future benefits of deferred tax assets will not be realized. Therefore, the Company established a full valuation allowance as of
December 31, 2021 and 2020. As of December 31, 2021 and 2020, the change in valuation allowance was $
The Company files income tax returns in the U.S. federal jurisdiction and various state jurisdictions, principally California and New Jersey. The Company is subject to examination by the various taxing authorities. The Company’s federal and state income tax returns for tax years beginning in 2018 remain subject to examination.
At
December 31, 2021 and 2020, the Company had approximately $
The Company’s gross R&D tax credits were approximately $
Coronavirus Aid, Relief, and Economic Security Act
On
March 27, 2020, the Coronavirus Aid, Relief, and Economic Security Act, (“CARES Act”), was enacted and signed into law. GAAP
requires recognition of the tax effects of new legislation during the reporting period that includes the enactment date. The CARES Act,
among other things, includes changes to the tax provisions that benefits business entities. It also makes certain technical corrections
to the 2017 Tax Cuts and Jobs Act and permits offsetting
F-14
CohBar, Inc.
Notes to Financial Statements
Note 10 – Stockholders’ Equity
Authorized Capital
The
Company has authorized the issuance and sale of up to
At-the-Market Offering
During
the year ended December 31, 2020, the Company entered into an At-the-Market Offering Sales Agreement (“ATM”) with
Virtu Americas, LLC as sales agent. During the year ended December 31, 2021, the Company sold
Underwritten Public Offerings
During
the year ended December 31, 2021, the Company completed an underwritten public offering of its securities (the “Public Offering”)
pursuant to which it sold
During
the year ended December 31, 2020, the Company completed an underwritten public offering of the Company’s securities (the
“Public Offering”) pursuant to which the Company sold
Stock Options
The
Company has an incentive stock plan, the Amended and Restated 2011 Equity Incentive Plan (the “2011 Plan”), and has
granted stock options to employees, non-employee directors and consultants from the 2011 Plan. Options granted under the 2011 Plan
may be Incentive Stock Options or Non-statutory Stock Options, as determined by the Administrator at the time of grant. During the
year ended December 31, 2020, the Company’s stockholders approved an amendment to the 2011 Plan to increase the number of
shares authorized for issuance under the 2011 Plan to a total of
During
the year ended December 31, 2021, the Company granted stock options to employees to purchase
In
connection with the appointment of Joseph Sarret as the Company’s Chief Executive Officer, the Company entered into an Inducement
Stock Option Agreement with Dr. Sarret on May 3, 2021.
F-15
CohBar, Inc.
Notes to Financial Statements
Note 10 – Stockholders’ Equity (continued)
During
the year ended December 31, 2021, stock options to purchase
During
the year ended December 31, 2021, stock options to purchase
During
the year ended December 31, 2020,
During
the year ended December 31, 2020, stock options to purchase
During
the year ended December 31, 2020, stock options to purchase
The Company recorded stock-based compensation as follows:
| For the Years Ended December 31, | ||||||||
| 2021 | 2020 | |||||||
| Research and development | $ | $ | ||||||
| General and administrative | ||||||||
| Total | $ | $ | ||||||
The following table represents stock option activity for the years ended December 31, 2021 and 2020:
| Weighted Average | Aggregate | |||||||||||||||||||||||||||
| Stock Options | Exercise Price | Fair Value | Contractual | Intrinsic | ||||||||||||||||||||||||
| Outstanding | Exercisable | Outstanding | Exercisable | Vested | Life (Years) | Value | ||||||||||||||||||||||
| Balance – January 1, 2020 | $ | $ | $ | $ | ||||||||||||||||||||||||
| Granted | - | |||||||||||||||||||||||||||
| Exercised | ( | ) | - | |||||||||||||||||||||||||
| Cancelled | ( | ) | - | |||||||||||||||||||||||||
| Balance – December 31, 2020 | $ | $ | $ | $ | ||||||||||||||||||||||||
| Granted | - | |||||||||||||||||||||||||||
| Exercised | ( | ) | - | |||||||||||||||||||||||||
| Cancelled | ( | ) | - | |||||||||||||||||||||||||
| Balance – December 31, 2021 | $ | $ | $ | $ | ||||||||||||||||||||||||
F-16
CohBar, Inc.
Notes to Financial Statements
Note 10 – Stockholders’ Equity (continued)
The following table summarizes information on stock options outstanding and exercisable as of December 31, 2021:
| Grant Price | Weighted Average Exercise | Total | Number | Weighted Average | |||||||||||||||||
| From | To | Price | Outstanding | Exercisable | Remaining Contractual Term | ||||||||||||||||
| $ | $ | $ | |||||||||||||||||||
| $ | $ | $ | |||||||||||||||||||
| $ | $ | $ | |||||||||||||||||||
Warrants
During
the year ended December 31, 2021, the Company granted warrants to two service providers to purchase a total of
During
the year ended December 31, 2021, warrants to purchase
During
the year ended December 31, 2021, warrants to purchase
During the year ended December 31, 2020, the Company issued warrants to purchase 10.6 million shares of the Company’s common stock as part of the Public Offering (see Note 10 – Underwritten Public Offerings) and to the note holders that extended the due date of their unsecured promissory notes (see Note 10 – Amendments to Notes and Warrants) and warrants to purchase 2.4 million shares of the Company’s common stock as part of the Private Offering that converted outstanding amounts due under the Company’s 8% Unsecured Promissory Notes due 2021 (see Note 7 - Notes Payable).
F-17
CohBar, Inc.
Notes to Financial Statements
Note 10 – Stockholders’ Equity (continued)
During
the year ended December 31, 2020, warrants to purchase
The following table represents warrant activity for the years ended December 31, 2021 and 2020:
| Weighted Average | Aggregate | |||||||||||||||||||||||||||
| Warrants | Exercise Price | Fair Value | Contractual | Intrinsic | ||||||||||||||||||||||||
| Outstanding | Exercisable | Outstanding | Exercisable | Vested | Life (Years) | Value | ||||||||||||||||||||||
| Balance – January 1, 2020 | $ | $ | $ | $ | ||||||||||||||||||||||||
| Granted | - | |||||||||||||||||||||||||||
| Exercised | ( | ) | - | |||||||||||||||||||||||||
| Cancelled | - | |||||||||||||||||||||||||||
| Balance – December 31, 2020 | $ | $ | $ | $ | ||||||||||||||||||||||||
| Granted | - | |||||||||||||||||||||||||||
| Exercised | ( | ) | - | |||||||||||||||||||||||||
| Cancelled | ( | ) | - | |||||||||||||||||||||||||
| Balance – December 31, 2021 | $ | $ | $ | $ | ||||||||||||||||||||||||
Amendments to Notes and Warrants
During the year ended December 31, 2020, the Company entered into amendments (the “Amendments”) with certain holders of the Company’s 8% Unsecured Promissory Notes (the “2018 Notes”) and Nontransferable Common Stock Purchase Warrants (the “2018 Warrants”). Pursuant to the Amendments, the maturity date of the applicable 2018 Notes was extended from March 29, 2021 to June 30, 2021 and the expiration date of the applicable 2018 Warrants was extended from March 29, 2021 to March 29, 2022. The terms of the applicable 2018 Notes were also amended to grant the holders of such 2018 Notes a right to participate in a future private offering of the Company’s securities upon terms substantially similar to those offered to investors in a future primary offering of the Company’s securities and to grant resale registration rights in connection therewith. The Company recognized $0.2 million of non-cash costs in Other Expenses in the accompanying statements of operations relating to the 2018 Warrants extension.
Also,
during the year ended December 31, 2020, the Company entered into amendments with certain holders of the Company’s Common Stock
Purchase Warrants (the “2017 Warrants”) pursuant to which the expiration date of the applicable 2017 Warrants was extended
from June 30, 2020 to September 30, 2021. The Company recognized $
The Company determined the proper
classification of the loan modification based on ASC 470-50, Debt Modifications and Extinguishments. Because the change in present value
of cash flows of the modified debt is less than
Employee Stock Purchase Plan
The
Company has an Employee Stock Purchase Plan (“ESPP”) in which employees may purchase shares with the amounts accumulated
during the offering period from employee directed payroll deferrals. Purchases of the Company’s common stock are equal to
F-18
CohBar, Inc.
Notes to Financial Statements
Note 11 – Non-Cash Expenses
The following table details the Company’s non-cash expenses included in the accompanying statements of operations:
| For the Years Ended December 31, | ||||||||
| 2021 | 2020 | |||||||
| Operating expenses: | ||||||||
| Stock-based compensation | $ | $ | ||||||
| Depreciation & amortization | ||||||||
| Subtotal | $ | $ | ||||||
| Other expense: | ||||||||
| Amortization of debt discount | ||||||||
| Equity modification | ||||||||
| Subtotal | $ | $ | ||||||
| Total non-cash expenses | $ | $ | ||||||
Note 12 – Subsequent Events
Management has evaluated subsequent events to determine if events or transactions occurring through the date on which the financial statements were issued require adjustment or disclosure in the Company’s financial statements.
Subsequent to December 31, 2021,
Subsequent
to December 31, 2021, the Company sold
Subsequent
to December 31, 2021, the Company repaid a promissory note, held by a director of the Company, totaling approximately $
F-19
Item 9. Changes in and Disagreements with Accountants on Accounting and Financial Disclosure
None.
Item 9A. Controls and Procedures
Evaluation of Disclosure Controls and Procedures
An evaluation was conducted under the supervision and with the participation of our management, including Joseph Sarret, our Chief Executive Officer, and Jeff Biunno, our Chief Financial Officer (collectively, the “Certifying Officers”), of the effectiveness of our disclosure controls and procedures as of December 31, 2021, as defined in Rules 13a-15(e) and 15d-15(e) of the Securities Exchange Act of 1934 (the “Exchange Act”). Based on that evaluation, our Chief Executive Officer and Chief Financial Officer concluded that, as of December 31, 2021, our disclosure controls and procedures were effective.
Management’s Report on Internal Control over Financial Reporting
Management is responsible for establishing and maintaining adequate internal control over financial reporting as such term is defined in Rule 13a-15(f) and 15(d)-15(f) under the Exchange Act. This rule defines internal control over financial reporting as a process designed by, or under the supervision of, Certifying Officers, to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with U.S. GAAP. Our internal control over financial reporting includes those policies and procedures that:
| ● | pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect the transactions and dispositions of our assets; |
| ● | provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with U.S. GAAP, and that receipts and expenditures are being made only in accordance with authorizations of our management and directors; and |
| ● | provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use or disposition of our assets that could have a material effect on the financial statements. |
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. In addition, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
Management’s Assessment
Our management, including our Chief Executive Officer and Chief Financial Officer, evaluated the effectiveness of our internal control over financial reporting based on the criteria established in Internal Control - Integrated Framework (2013), issued by the Committee of Sponsoring Organizations of the Treadway Commission. Our management, including our Chief Executive Officer and Chief Financial Officer, have concluded that as of December 31, 2021, our internal control over financial reporting was effective.
We have limited capital resources and have given priority in the use of those resources to our research and development efforts. If we are unable to maintain effective internal control over financial reporting, we may not be able to report our financial results accurately, prevent fraud or file our periodic reports in a timely manner. We continue to evaluate the effectiveness of our internal controls and procedures on an on-going basis. As our operations continue to grow and become more complex, we intend to hire additional personnel in financial reporting and other areas.
Auditor Attestation
This Annual Report on Form 10-K does not include an attestation of our registered public accounting firm regarding internal control over financial reporting. Management’s report was not subject to attestation by our registered public accounting firm pursuant to applicable rules of the Securities and Exchange Commission.
Changes in Internal Control over Financial Reporting
There were no changes in our internal control over financial reporting during the last fiscal quarter that have materially affected, or are reasonably likely to materially affect, our internal control over financial reporting.
Item 9B. Other Information
None.
Item 9C. Disclosure Regarding Foreign Jurisdictions that Prevent Inspections
None.
44
PART III
Item 10. Directors, Executive Officers and Corporate Governance
The information required by this item is incorporated herein by reference to our Proxy Statement with respect to our 2022 Annual Meeting of Stockholders to be filed with the SEC within 120 days of the end of the fiscal year covered by this Annual Report on Form 10-K, or will be included in an amendment to this Annual Report on Form 10-K.
Item 11. Executive Compensation
The information required by this item is incorporated herein by reference to our Proxy Statement with respect to our 2022 Annual Meeting of Stockholders to be filed with the SEC within 120 days of the end of the fiscal year covered by this Annual Report on Form 10-K, or will be included in an amendment to this Annual Report on Form 10-K.
Item 12. Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters
Equity Compensation Plan Information
The following table provides information about our equity compensation plan as of December 31, 2021:
| Number of securities to be issued upon exercise of options warrants and rights | Weighted-average exercise price of outstanding options warrants and rights | Number of securities remaining available for future issuance under equity compensation plans (excluding securities reflected in column (a)) | ||||||||||
| Plan Category | (a) | (b) | (c) | |||||||||
| Equity compensation plans approved by stockholders | 8,092,335 | $ | 1.84 | 3,726,857 | (2) | |||||||
| Equity compensation plans not approved by stockholders | 4,552,671 | (1) | $ | 1.18 | - | |||||||
| Total | 12,645,006 | $ | 1.60 | 3,726,857 | ||||||||
| (1) | Consists of inducement stock options granted to our Chief Executive Officer pursuant to an employment agreement, warrants issued to our former Chief Operating Officer pursuant to an employment agreement, warrants issued to four consultants pursuant to consulting agreements, and warrants issued to the Alzheimer’s Drug Discovery Foundation for the 2013 grant. |
| (2) | Consists of securities for two equity compensation plans approved by the Company’s stockholders, (i) an incentive stock plan, the Amended and Restated 2011 Equity Incentive Plan, as amended (the “2011 Plan”), which the Company has granted stock options to employees, non-employee directors and consultants; and (ii) an Employee Stock Purchase Plan which allows employees of the Company to purchase shares through payroll deductions during set offering periods. |
Beneficial Ownership
The information required by this item is incorporated herein by reference to our Proxy Statement with respect to our 2022 Annual Meeting of Stockholders to be filed with the SEC within 120 days of the end of the fiscal year covered by this Annual Report on Form 10-K, or will be included in an amendment to this Annual Report on Form 10-K.
Item 13. Certain Relationships and Related Transactions, and Director Independence
The information required by this item is incorporated herein by reference to our Proxy Statement with respect to our 2022 Annual Meeting of Stockholders to be filed with the SEC within 120 days of the end of the fiscal year covered by this Annual Report on Form 10-K, or will be included in an amendment to this Annual Report on Form 10-K.
Item 14. Principal Accounting Fees and Services
The information required by this item is incorporated herein by reference to our Proxy Statement with respect to our 2022 Annual Meeting of Stockholders to be filed with the SEC within 120 days of the end of the fiscal year covered by this Annual Report on Form 10-K, or will be included in an amendment to this Annual Report on Form 10-K.
45
PART IV
Item 15. Exhibits, Financial Statement Schedules
The financial statements, together with the report thereon of Marcum LLP, are included on the pages indicated below:
Financial Statements and Schedules
Financial statement schedules have been omitted because they are not applicable or the required information is shown in the financial statements or notes thereto.
Exhibits
The following exhibits are filed herewith and this list is intended to constitute the exhibit index.
46
47
48
| * | Indicates management contract, compensatory agreement or arrangement, in which our directors or executive officers may participate. |
Item 16. Form 10-K Summary
Not applicable.
49
SIGNATURES
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, the registrant has duly caused this report to be signed on its behalf by the undersigned, thereunto duly authorized.
| Date: March 29, 2022 | COHBAR, INC. | |
| By: | /s/ Jeffrey F. Biunno | |
| Jeffrey F. Biunno | ||
Chief Financial Officer | ||
| (Principal Financial and Accounting Officer) | ||
POWER OF ATTORNEY
Each person whose individual signature appears below hereby authorizes and appoints Jeffrey F. Biunno and Joseph Sarret, and each of them, with full power of substitution and resubstitution and full power to act without the other, as his true and lawful attorney-in-fact and agent to act in his name, place and stead and to execute in the name and on behalf of each person, individually and in each capacity stated below, and to file any and all amendments to this report, with all exhibits thereto, and other documents in connection therewith, with the Securities and Exchange Commission, granting unto said attorneys-in-fact and agents, and each of them, full power and authority to do and perform each and every act and thing, ratifying and confirming all that said attorneys-in-fact and agents or any of them or their or his substitute or substitutes may lawfully do or cause to be done by virtue thereof.
Pursuant to the requirements of the Securities Exchange Act of 1934, this report has been signed below by the following persons on behalf of the registrant and in the capacities and on the dates indicated.
| Signature | Title | Date | ||
| /s/ Joseph Sarret | Chief Executive Officer and Director |
March 29, 2022 | ||
| Joseph Sarret | (Principal Executive Officer) | |||
| /s/ Jeffrey F. Biunno | Chief Financial Officer, Treasurer and Secretary | March 29, 2022 | ||
| Jeffrey F. Biunno | (Principal Financial and Accounting Officer) | |||
| /s/ David Greenwood | Chairman of the Board of Directors | March 29, 2022 | ||
| David Greenwood | ||||
| /s/ Carol Nast | Director | March 29, 2022 | ||
| Carol Nast | ||||
| /s/ Joanne Yun | Director | March 29, 2022 | ||
| Joanne Yun | ||||
/s/ Phyllis Gardner |
Director |
March 29, 2022 | ||
| Phyllis Gardner | ||||
/s/ Albion J. Fitzgerald |
Director |
March 29, 2022 | ||
| Albion J. Fitzgerald | ||||
/s/ Misha Petkevich |
Director |
March 29, 2022 | ||
| Misha Petkevich |
50